FROM OUR 2019 SCIENCE EARLY CAREER PRIZE LECTURER
Genome-wide association studies (GWAS) have identified over 300 loci in the human genome harbouring genetic variants that alter type 2 diabetes (T2D) risk.1,2 In the last decade, it has become clear that T2D genetic risk variants are enriched within transcriptional enhancers that are active in pancreatic islets,3–5 indicating that T2D genetic risk is underlined by defects in islet gene regulation.
Transcriptional enhancers can regulate one or more genes at varying distances and irrespective of their relative position (i.e. they can regulate genes either up- or downstream of them). In practice, the complex nature of transcriptional enhancers has hampered the identification of true diabetes risk effector genes and the translation of T2D GWAS variants into targeted therapies.
To address this problem, we applied promoter capture Hi-C6 to create a genome-wide map of promoter–enhancer interactions in adult human pancreatic islets.7
Using this approach, we identified target genes for approximately 80% of all active human islet enhancers (36,664 of 45,683 enhancers). More specifically, this map enabled the systematic identification of genes that are regulated by enhancers carrying T2D risk variants. We observed that T2D variants often interact with more than one gene and that, unlike what has been assumed until now, the nearest genes are not always the true targets of T2D susceptibility variants. Indeed, over 75% of loci with at least one T2D risk variant in an islet enhancer were assigned to novel target genes.
These results have major implications for the interpretation of past and future genetic signals identified in GWAS, as it is often assumed that GWAS variants are flagging the nearest gene. By bypassing the genome’s linear representation which is more commonly used, and putting genetic risk loci in the context of the genome’s 3D conformation, we have gained access to a collection of new putative diabetes risk effector genes.
We decided to validate our in silico predictions using CRISPR-Cas9 perturbations in a human pancreatic β cell in vitro model. To this end, we first inspected the epigenomic landscape of the human pancreatic β cell line EndoC-βH38 by performing an assay for transposase-accessible chromatin using sequencing (ATAC-seq9) to profile all accessible chromatin regions in the genome, a proxy for active and regulatory sequences. Our analysis revealed that EndoC-βH3 cells recapitulate the epigenomic landscape of human pancreatic islets at key loci, including those implicated in T2D genetic risk.</p
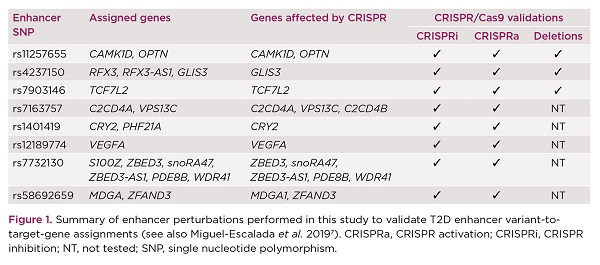
With these reassuring results, we set out to validate the in silico predictions at ten specific islet enhancers that harbour T2D risk variants, by performing epigenomic modulation of the target enhancers and/or by deleting them (Figure 1). Epigenomic modulation was achieved by targeting T2D enhancers with a catalytically inactive Cas9 (dCas9) tethered with specific effector domains (VP64-P65-HSF1 for CRISPR activation and KRAB for CRISPR inhibition). In comparison, deletion of islet enhancers was achieved by co-expressing Cas9 with two single guide RNAs flanking the target enhancers.
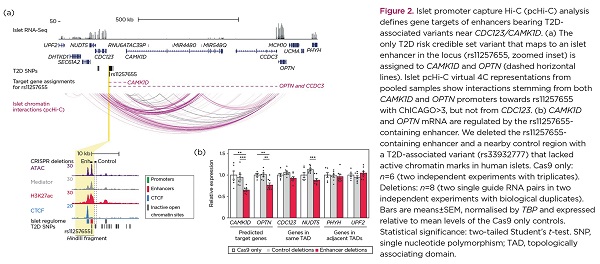
One of the investigated T2D loci was CDC123/CAMK1D, where a single risk variant resides in an islet enhancer with classic enhancer features (e.g. high chromatin accessibility and enrichment for Mediator and H3K27ac). Previously, human islet eQTL studies had prioritised CAMK1D as the target of this enhancer variant.10 Nevertheless, our analysis by promoter capture Hi-C also enabled detection of a very long range interaction with a gene that has not been previously implicated in diabetes, OPTN, located >800kb from the risk variant (Figure 2a). Deletion of this T2D enhancer in EndoC-βH3 cells led to marked downregulation of both CAMK1D and OPTN, but not of the nearest gene, CDC123 (Figure 2b). Further supporting our in silico assignments, CRISPR-mediated activation of this T2D enhancer resulted in overexpression of CAMK1D and OPTN, whereas CRISPR-mediated inhibition led to an opposite effect.
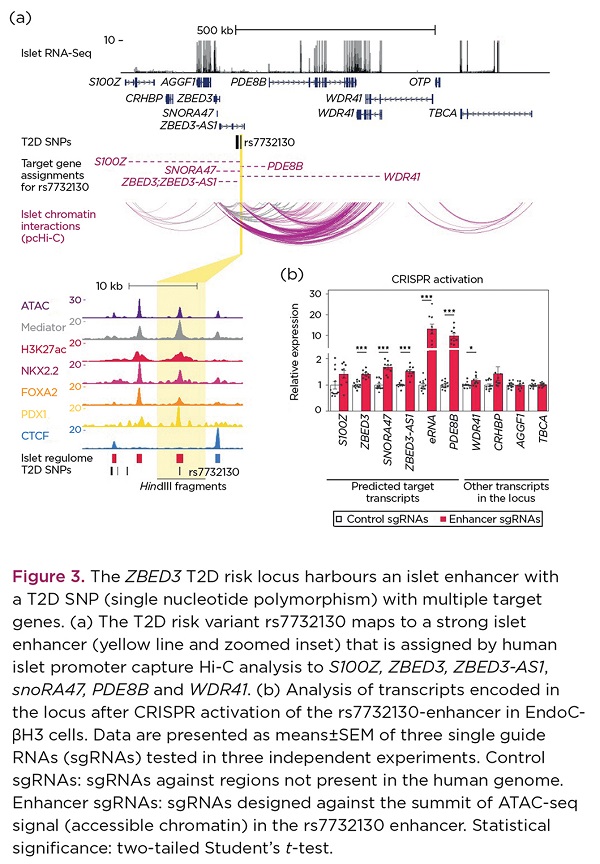
At the ZBED3 T2D locus, we observed a remarkable connectivity between the T2D enhancer and multiple genes in the locus (Figure 3a), which was corroborated by the CRISPR perturbation experiments in EndoC-βH3 cells (Figure 3b). Altogether, our experimental validations demonstrated that the detected chromatin enhancer–promoter interactions reflect functional chromatin interactions in human pancreatic islets.
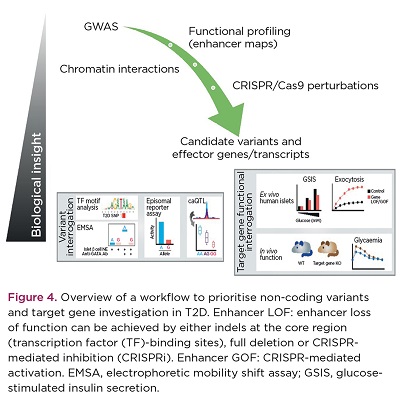
This study has revealed 3D chromatin architecture analysis coupled with genome editing as a powerful framework for interpretation of T2D genetic association signals (Figure 4). Furthermore, the results shed light into unexpected regulatory links that may be affected by T2D susceptibility variants, bringing to our attention new potential players in T2D aetiology. Future functional investigation of these new T2D genes should illuminate their contribution to T2D risk processes.
INÊS CEBOLA
Department of Metabolism, Digestion and Reproduction, Imperial College London
REFERENCES
- Mahajan A et al. 2018 Nature Genetics 50 1505–1513.
- Spracklen CN et al. 2020 Nature 582 240–245.
- Parker SCJ et al. 2013 Proceedings of the National Academy of Sciences of the USA 110 17921–17926.
- Pasquali L et al. 2014 Nature Genetics 46 136–143.
- Cebola I 2019 Current Diabetes Reports 19 145.
- Javierre BM et al. 2016 Cell 167 1369–1384.e19.
- Miguel-Escalada I et al. 2019 Nature Genetics 51 1137–1148.
- Benazra M et al. 2015 Molecular Metabolism 4 916–925.
- Buenrostro JD et al. 2013 Nature Methods 10 1213–1218.
- van de Bunt M et al. 2015 PLoS Genetics 11 e1005694.




{kind=link}
{kind=link}
{kind=link}
{kind=link}