
Kerri Devine and Rebecca M Reynolds
Epidemiological research carried out in the late 1980s and 1990s revealed a link between low birthweight and the onset of metabolic disorders, including obesity, hypertension and type 2 diabetes, in adulthood.
To explain this observation, in 1993, David Barker proposed that a fetus exposed to certain adverse factors during critical points in its development will make adaptations which result not only in restricted growth (and hence small birth size), but in permanent physiological changes, for example to blood vessel or endocrine functions.1 These changes may have a survival advantage for the fetus, but are harmful in the long term, hence linking the early life environment with adult disease.
Barker believed this ‘developmental programming’ was driven by poor maternal nutrition, but decades of subsequent animal and observational studies have since expanded the potential underlying mechanisms to include a range of preconceptual, periconceptual and other intrauterine exposures. Excessive exposure to glucocorticoids in the womb is proposed as a key underpinning mechanism.
GLUCOCORTICOIDS IN PREGNANCY
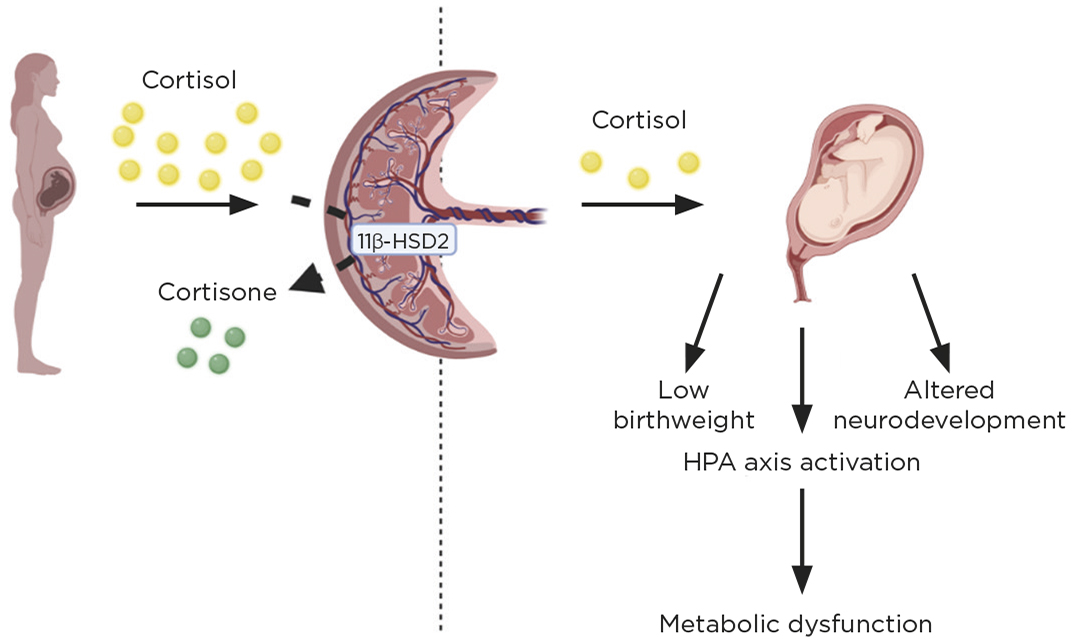
Levels of cortisol – the primary stress hormone produced by the adrenal gland – typically rise threefold in the human mother during pregnancy. The fetus is usually protected from this steroid surge by the actions of a placental enzyme (called 11β-hydroxysteroid dehydrogenase type 2, or 11β-HSD2) which inactivates glucocorticoids. Consequently only 10–20% of maternal cortisol transfers.
A rise in maternal cortisol in response to ‘stress’, or any small reduction in enzyme function, would result in a significant increase in circulating fetal glucocorticoid.

Maternal blood cortisol (yellow circles) reaches the placenta where it is partially metabolised by 11β-HSD2 into the inactive cortisone (green circles). An increase in cortisol transfer to the fetus (due to increased maternal levels, or reduced inactivation) results in low birthweight, long term activation of the HPA and alterations in development of the limbic system. This links the in utero environment with adult health and disease. Figure created with BioRender.com
IMPACT OF HIGH GLUCOCORTICOID LEVELS
The ‘glucocorticoid hypothesis’ of fetal developmental programming was conceived when rodents lacking this enzyme (and therefore with high glucocorticoid transfer across the placenta) were noted to have smaller offspring.2 Similarly, humans with lower placental 11β-HSD2 function, and those with the very rare homozygous 11β-HSD2 mutation, have lower birthweights than siblings. Further animal studies confirmed that greater placental steroid transfer (either due to maternal steroid administration or inhibition of 11β-HSD2) resulted in offspring that were not only smaller, but also, importantly, developed hypertension, altered fat metabolism and other features of the ‘low birthweight baby’ phenotype.
MECHANISM OF ACTION
This enhanced glucocorticoid exposure during development appears to have long term effects on glucocorticoid regulation. The plasma concentration of glucocorticoids is controlled by the hypothalamic-pituitary-adrenal (HPA) axis, and low birthweight animals and humans have a change in the ‘sensitivity’ of the axis towards higher glucocorticoid concentrations. Rats which have been exposed to steroids in utero have high basal glucocorticoid levels throughout life, for example.
Maternal liquorice consumption in pregnancy has been used as a novel marker of fetal cortisol exposure, since liquorice contains a natural 11β-HSD2 inhibitor (glycyrrhizin). Children whose mothers had high liquorice intake during pregnancy, compared with those with none, had greater and brisker salivary cortisol production on waking.3 Long term population cohort studies have demonstrated that those with lower birth weights have higher cortisol concentrations, as well as enhanced cortisol production to stimulus, in early and late adulthood. This again suggests a lifelong alteration to the axis.
It is intuitive to endocrinologists that dysregulation of the HPA axis results in hypertension, obesity, insulin resistance and cardiovascular disease, as this is typical of Cushing’s syndrome. We have already demonstrated the principle that greater fasting cortisol in humans is associated with a variety of adverse metabolic features, including hypertension, hyperglycaemia and dyslipidaemia.4 Furthermore, in cohort and cross-over studies, we have linked these adverse features with both low birthweight and HPA axis activation.5 Children whose mothers were treated with glucocorticoids during pregnancy have higher blood pressure and insulin resistance in adolescence and early adulthood.6,7
OVERCOMING CHALLENGES IN ANALYSIS
One of the key limitations in this field results from the challenges of measuring cortisol exposure in pregnancy, with many factors, such as time of sample collection and several maternal characteristics, leading to large variations in measurements of cortisol in blood during pregnancy.8 One potential way to overcome this is through measurement of cortisol in hair, with 1cm of hair representing a month of cortisol exposure. We measured cortisol in maternal hair samples collected at the time of birth, representing cortisol exposure in the last three months of pregnancy.9 The newborn babies underwent brain scans using magnetic resonance imaging (MRI) to examine the structure and connections of the amygdala, the part of the brain that processes emotions. We found that being exposed to higher cortisol levels in the womb affected babies in different ways based on their sex: boys showed alterations in the fine structure of their amygdala, while girls displayed changes in the way that that brain region connected to other neural networks. Thus, cortisol exposure in the womb also appears important for offspring neurodevelopment.
A further limitation is the observational nature of the available data, and the challenge of demonstrating a causative role for glucocorticoids in human studies. The Early Growth Genetics (EGG) Consortium has made major advances using advanced statistical methods (Mendelian randomisation) to demonstrate how maternal and fetal genetic effects may explain some of the observed associations between size at birth and adult diseases, including hypertension and type 2 diabetes. The knowledge that morning cortisol is genetically determined10 paves the way for further exploration of the ‘glucocorticoid hypothesis’.
KERRI DEVINE and REBECCA M REYNOLDS
Clinical Research Fellow and Professor of Metabolic Medicine, Centre for Cardiovascular Science, Queen’s Medical Research Institute, Edinburgh
References
Barker DJ et al. 1993 Lancet 341 938–941.
Edwards CR et al. 1993 Lancet 341 355–357.
Räikkönen K et al. 2010 Psychoneuroendocrinology 35 1587–1593.
Reynolds RM et al. 2010 Journal of Clinical Endocrinology & Metabolism 95 1602–1608.
Reynolds RM et al. 2001 Journal of Clinical Endocrinology & Metabolism 86 245–250.
Doyle LW et al. 2000 Clinical Science 98 137–142.
Dalziel SR et al. 2005 Lancet 365 1856–1862.
Bleker LS et al. 2017 Psychoneuroendocrinology 83 172–181.
Stoye DQ et al. 2020 Elife 9 e60729.
Crawford AA et al. 2019 European Journal of Endocrinology 181 429–438.



{kind=link}