Primary congenital hypothyroidism (CH) is the most common neonatal endocrine defect, and delayed diagnosis can result in significant neurodevelopmental impairment. The UK CH screening programme, which detects CH using a neonatal blood spot thyrotropin (TSH) measurement, has been resoundingly successful in negating requirements for special schooling for children with CH. However some CH patients may still exhibit subtle cognitive deficiencies.1
Thyroid dysgenesis results in CH, due to complete failure of thyroid development (agenesis), ectopic development or (rarely) development of a hypoplastic but normally located thyroid gland. CH resulting from dyshormonogenesis may be goitrous, and occurs due to a specific defect in one of the components of the thyroid hormone biosynthetic machinery. Older studies, using TSH screening cut-offs of >20mU/l, attributed at least 80% of CH to thyroid dysgenesis. However, more recent studies, using lower TSH screening cut-offs, have found that most CH cases have a normally located thyroid gland in situ.2,3
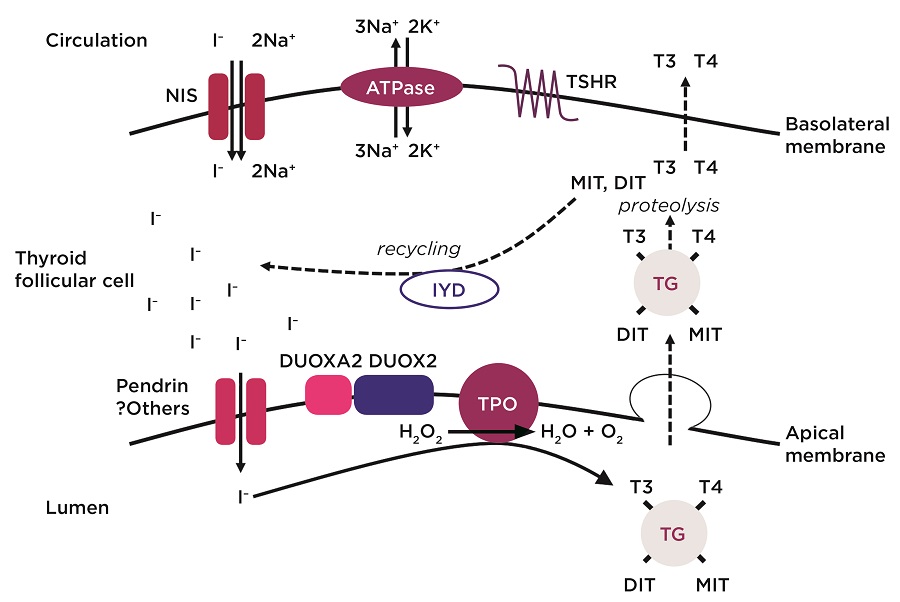
The figure above outlines the key molecules involved in thyroid hormone biosynthesis.
NORMAL DEVELOPMENT AND HORMONE BIOSYNTHESIS
Our understanding of normal thyroid physiology and development has been key to identifying genetic causes of CH.
During embryogenesis, a thyroid domain is specified in the foregut endoderm, close to the tongue base. Following evagination of a thyroid primordium, thyroid precursor cells migrate downwards, reaching their final position anterior to the trachea around 40 days post fertilisation. Terminal differentiation then begins with polarisation and adhesion of individual thyroid follicular cells to form functional follicles. Fetal thyroid hormones are detectable in the circulation around gestational week 12.
GENES KNOWN TO BE IMPLICATED IN CH
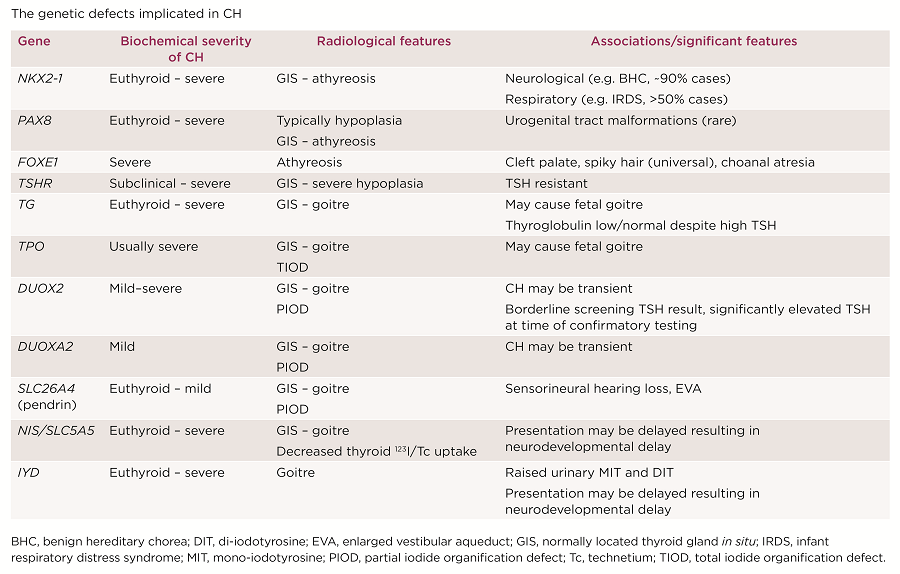
The majority of dyshormonogenesis is explained by mutations in key genes encoding components of the thyroid hormone biosynthesis pathway (Figure). Recessively inherited mutations in any of these molecules and dominantly inherited DUOX2, IYD and DUOXA2 mutations have all been associated with CH or goitre (Table).
Four key thyroid transcription factors mediate normal thyroid development: NKX2-1, PAX8, FOXE1 and HHEX. Their combined, co-ordinated expression defines developing thyroid follicular cells.4 Heterozygous NKX2-1 and PAX8 mutations and recessively inherited FOXE1 mutations are all associated with thyroid dysgenesis, and may also result in extrathyroidal phenotypes, reflecting individual roles of these transcription factors in the morphogenesis of additional organs (Table). Mutations in these genes are rare, accounting for <5% of CH (Table).2 Mono- or biallelic mutations in the TSHR gene cause a variable spectrum of TSH resistance, ranging from thyroid hypoplasia and severe CH to mild isolated hyperthyrotrophinaemia, with normal thyroid morphology. TSHR mutations are relatively common, accounting for up to ~20% of paediatric non-autoimmune hyperthyrotrophinaemia (Table).5
Collectively, <20% of CH is accounted for by known genetic causes, and the frequent sporadic occurrence of thyroid dysgenesis and high discordance rate in monozygotic twins has led to the hypothesis that thyroidal somatic mutations account for thyroid dysgenesis. However, observations that 2% of thyroid dysgenesis is familial, and the enrichment of extrathyroidal developmental abnormalities in CH cases, support the existence of hitherto undiscovered genetic causes.2

The genetic defects implicated in CH
WHEN IS GENETIC ASCERTAINMENT IMPORTANT?
On receipt of a positive CH screening test result, parents invariably ask why their baby has CH. A confirmatory genetic diagnosis may answer this question, and facilitate genetic counselling and management of cases likely to exhibit syndromic features.
It may enable cases of likely transient CH to be identified (usually due to DUOX2 or DUOXA2 mutations), informing a trial of levothyroxine withdrawal. Additionally, CH with certain genetic aetiologies (NIS, IYD, DUOX2 mutations) may present with normal neonatal TSH levels but evolve to significant hypothyroidism in later childhood. A falsely reassuring CH screening result in such cases may lead to delayed diagnosis with adverse sequelae. However, genetic ascertainment may promote early biochemical surveillance of affected siblings.6,7 TSHR mutations may cause resistance to TSH, resulting in persistently elevated TSH despite high/high normal free thyroxine. However, levothyroxine treatment may not be required for normal growth and development, and attempts to normalise TSH may provoke thyrotoxic symptoms. Detection of a TSHR mutation in such cases may therefore aid clinical management by permitting a more appropriate (higher) TSH target range to be agreed.8
LESSONS FROM NEXT GENERATION SEQUENCING
Next generation sequencing technologies have demonstrated a role for oligogenicity in the pathogenesis of CH, which may explain the variable penetrance observed.9,10 Whole exome sequencing in familial cases has identified mutations in borealin (CDCA8) as a novel monogenic cause of thyroid dysgenesis.11 Additionally, association of mutations in known genes with unexpected thyroid morphology (e.g. pendrin (SLC26A4) in thyroid dysgenesis) has broadened our perspective on the phenotypes associated with particular genetic mutations.12 It remains to be seen whether further studies, including those using whole genome sequencing, will identify additional genetic causes in known or novel candidate genes.
Nadia Schoenmakers, Wellcome Intermediate Clinical Fellow & Honorary Consultant Endocrinologist, University of Cambridge Metabolic Research Laboratories, Wellcome Trust-MRC Institute of Metabolic Science, Addenbrooke’s Hospital, Cambridge
REFERENCES
- Rovet JF 2005 Pediatrics 115 e52–e57.
- Szinnai G 2014 Endocrine Development 26 60–78.
- Corbetta C et al. 2009 Clinical Endocrinology 71 739–745.
- Nilsson M & Fagman H 2017 Development 144 2123–2140.
- Cassio A et al. 2013 Journal of Clinical Research in Pediatric Endocrinology 5 Suppl 1 29–39.
- Spitzweg C & Morris JC 2010 Molecular & Cellular Endocrinology 322 56–63.
- Moreno JC et al. 2008 New England Journal of Medicine 358 1811–1818.
- Clifton-Bligh RJ et al. 1997 Journal of Clinical Endocrinology & Metabolism 82 1094–1100.
- de Filippis T et al. 2017 Human Molecular Genetics 26 2507–2514.
- Aycan Z et al. 2017 Journal of Clinical Endocrinology & Metabolism doi: 10.1210/jc.2017-00529. 11. Carré A et al. 2017 Human Molecular Genetics 26 599–610.
- Kühnen et al. 2014 Journal of Clinical Endocrinology & Metabolism 99 E169–E176.




{kind=link}
{kind=link}