We think of diabetes pathophysiology as primarily driven by insulin insufficiency. However, long before diagnosis of type 2 diabetes, a profound excess of insulin predicts the people with obesity and pre-diabetes who will progress to disease.1 Moreover, genetic evidence suggests a very early excess of insulin production prior to type 1 diabetes.2,3 Non-trivial persistent endogenous insulin secretion may shape the course of type 1 diabetes,4 a disease we used to define by the complete absence of endogenous insulin.
Here, we discuss parallels in the complex relationship between insulin and both major forms of diabetes. We describe pharmacological and nonpharmaceutical methods, including dietary modifications, by which insulin can be modulated for the prevention and improved management of diabetes.

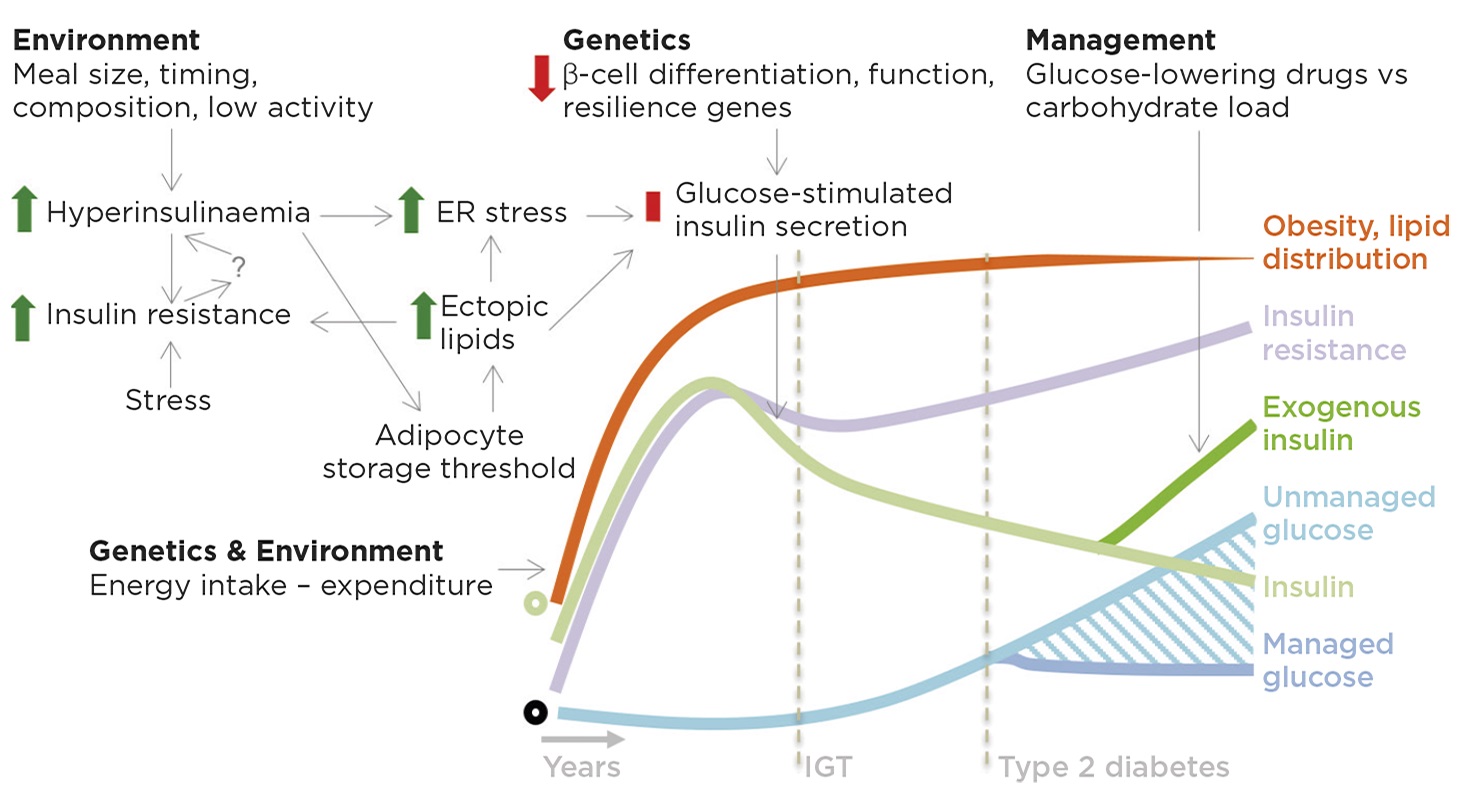
Figure 1. Roles for insulin in insulin resistance, obesity and type 2 diabetes. On a background of genetic and environmental factors that increase food intake, we propose that meal size, timing and macronutrient composition stimulate excess insulin production/secretion in fasting and fed states. Hyperinsulinaemia may contribute to insulin resistance through receptor and post-receptor desensitisation, possibly further promoting hyperinsulinaemia via unknown mechanisms. Hyperinsulinaemia drives lipid storage in adipocytes, which eventually leads to adipocyte dysfunction and lipid spillover into other tissues. Ectopic lipids are deposited in many tissues including the pancreas where, together with the increased insulin demand, they produce endoplasmic reticulum (ER) stress and metabolic dysfunction, resulting in impaired glucose-stimulated insulin secretion. Impaired insulin secretion in response to glucose begets impaired glucose tolerance (IGT) and eventually type 2 diabetes. The late stages of type 2 diabetes are associated with more severe changes in β-cell differentiation state and cell death. Type 2 diabetes management is a balance between glucose-lowering drugs, including insulin, and glucose load, which can be modified by diet. Green circle denotes normal insulin; black circle represents the norm for other features. © J D Johnson & J A Kushner
OBESITY, INSULIN RESISTANCE AND TYPE 2 DIABETES
The coincidental timing of hyperinsulinaemia, insulin resistance and obesity has led to a tripartite ‘chicken and egg and egg salad’ problem. One cannot assign causality from correlational clinical studies, even ones with very sensitive measures aimed at determining which of these features can be detected first.
Pharmacological reduction of insulin with diazoxide or octreotide caused weight loss in some clinical trials, suggesting that excess insulin plays a causal role in human obesity,5,6 but both drugs have multiple effects besides insulin inhibition. Animal models wherein insulin production can be suppressed without impairing long term glucose homeostasis provide robust evidence that hyperinsulinaemia is a biological requirement for diet-induced obesity7 and a partial driver of age-dependent insulin sensitivity.8 Modest hyperinsulinaemia is also sufficient for adipose tissue inflammation.9 Thus, factors that contribute to excess insulin production may predispose individuals to later type 2 diabetes.
In the majority of people, dietary carbohydrates are the major stimulus for insulin secretion. Diets that are high in refined carbohydrates are now globally ubiquitous from a very young age. Epidemiological studies aimed at determining whether high carbohydrate diets promote obesity, insulin resistance and diabetes are fraught with confounders, but the rise in consumption of sugars and other carbohydrates is hard to ignore. Long term interventional diet studies, especially in children, will be required to determine the impact of dietary macronutrients on obesity and the progressions to pre-diabetes and diabetes.
Similarly, fetal and early childhood environmental programming due to excess β-cell stimulation may lead to β-cell dysfunction and increased β-cell mass,7 an initially adaptive feature that eventually promotes hyperinsulinaemia and then insulin resistance. In addition, genome-wide association studies indicate that genes regulating the development of functional β-cell mass mediate susceptibility to type 2 diabetes.10 Collectively, a picture is emerging wherein abnormal β-cell function, first with an excess insulin release and then via impaired β-cell responsiveness, plays a key role in the progression from obesity, through impaired glucose tolerance, and into frank type 2 diabetes (Figure 1).
MANAGEMENT OF TYPE 2 DIABETES
At current rates, a majority of patients with type 2 diabetes will eventually be prescribed exogenous insulin. Recombinant insulin drugs have increasingly sophisticated pharmacokinetics but they cannot permanently reverse the course of the disease, even when administered early to promote ‘β-cell rest’.11 We speculate that this is probably because prolonged elevation of exogenous insulin causes modest but consistent weight gain,12 or is weight neutral at best. Some glucose transport inhibitors or GLP-1 (glucagon-like peptide-1) agonists (both of which may alter macronutrient metabolism and lower fasting insulin) appear to have superior cardiovascular benefits compared with long-acting insulins.12–14
‘Balanced’ diet and other lifestyle modifications continue to be advanced as first-line therapies for type 2 diabetes, despite evidence that traditional lifestyle approaches do not reliably or robustly alter the course of disease.15 People are encouraged by government agencies to achieve a negative energy balance by increasing exercise and reducing calorie intake, but the environment in which we live makes this nearly impossible for most, and clinical evidence in favour of this approach is lacking.
Yet, we know from recently published work that type 2 diabetes remission is possible with extreme caloric restriction (below 850kcal/day).16 Similarly, new research is continuing to provide evidence of ways to tailor eating habits that do not involve such stringent calorie restriction. For example, maintaining food intake within a shorter than normal window can reduce insulin secretion and improve apparent insulin sensitivity in men with prediabetes.17
The scientific literature and the public sphere remain unsettled as to which dietary modifications are best for healthy populations or people with diabetes. Dietary guidelines have stressed a reduction in saturated fat consumption (www.diabetes.org.uk), while others have argued from a biological standpoint that people with diabetes are carbohydrate-intolerant, which has led to efforts to prevent and treat type 2 diabetes with low carbohydrate diets.18

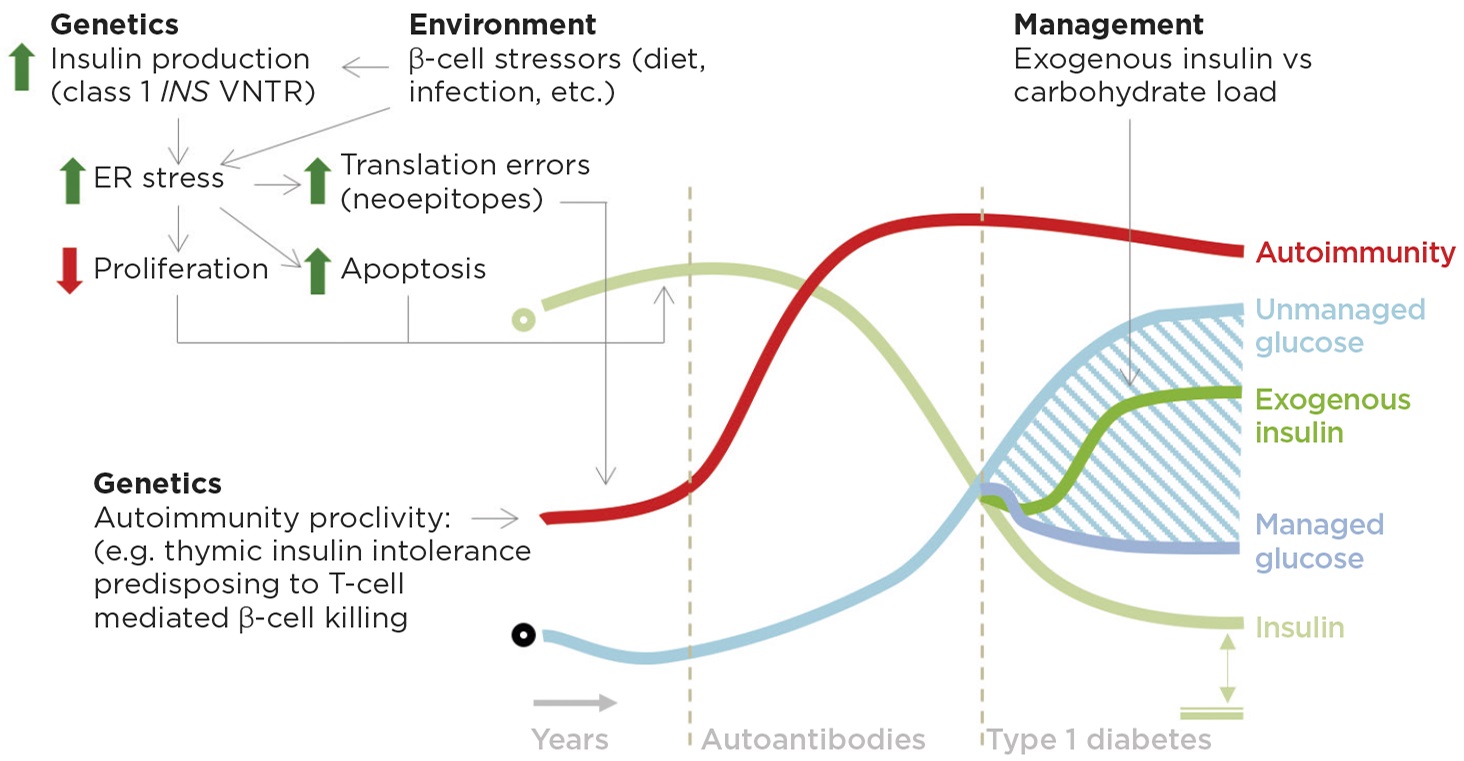
Figure 2. Potential role of insulin in autoimmunity and type 1 diabetes. On a background of genetic autoimmunity susceptibility, increased insulin production driven in part by at-risk VNTR (variable number tandem repeat) alleles and, we speculate, in part by diet leads to a cell autonomous stress on β-cell insulin production capacity that increases the chances of errors and decreases β-cell proliferation in a critical window for the development of thymic tolerance and insulin secretion capacity respectively. Type 1 diabetes management should consider carbohydrate load, exogenous insulin and residual endogenous insulin (indicated by double-headed arrow to double line, bottom right). ER, endoplasmic reticulum; green circle denotes normal insulin; black circle represents the norm for other features. © J D Johnson & J A Kushner
Though not a randomised trial, impressive results including 12% weight loss and diabetes remission were found in a cohort of patients consuming a low carbohydrate, ketogenic diet with supportive coaching.19 Similarly, a small randomised trial recently found that low carbohydrate, high fat nutrition was associated with reduced medication use and improved secondary outcome measures in patients with type 2 diabetes.20
Together, these results support the hypothesis of low carbohydrate nutrition for type 2 diabetes and reinforce the need for well-funded, highly powered randomised trials in large populations.
INITIATION AND PROGRESSION OF TYPE 1 DIABETES
While the genetics of type 1 diabetes point squarely at the immune system as the largest arbiter of risk, variation upstream of the human insulin gene is the second most important genetic factor. At-risk INS alleles decrease insulin production in the thymus, potentially impairing immune tolerance to insulin,21 but simultaneously increase insulin production in β-cells by approximately 30%.2,21 Increased insulin production would be predicted to increase β-cell stress and decrease β-cell proliferation.22 Similarly, mutations in GATA4, a transcription factor required for normal insulin processing, predispose to type 1 diabetes.23 An early increase in β-cell stress, perhaps in combination with β-cell agitations from diet,24 obesity,25 or other environmental factors, could promote the production of disease initiating neoepitopes26 (Figure 2).
Studies are underway to test the hypothesis that an early increase in insulin production contributes to the triggering and/or progression or type 1 diabetes in immunologically susceptible mice. Controlled human trials of diets meant to minimise β-cell stress are warranted in children at risk for type 1 diabetes.
EXOGENOUS AND ENDOGENOUS INSULIN IN TYPE 1 DIABETES
People with type 1 diabetes have a fundamental inability to properly dispose of carbohydrates and therefore have uncontrolled fluctuations in blood glucose. Hyperglycaemia causes severe, but reversible, impairment of remaining β-cells27 and promotes diabetes complications.28
There are essentially two ways to dampen harmful glucose excursions: (a) moderate glucose intake, or (b) enhance glucose disposal, either via the insulin system, or independently (e.g. SGLT (sodium–glucose co-transporter) inhibitors). A recent study has demonstrated the feasibility of using very low carbohydrate diets to prevent glucose excursions in children and adults.29 We look forward to robust randomised trials to assess the generalisability of this approach to the wider population of people with type 1 diabetes.
James D Johnson, Diabetes Research Group, Department of Cellular and Physiological Sciences, University of British Columbia and Institute for Personalized Therapeutic Nutrition, Vancouver, BC, Canada
Jake A Kushner, McNair Interests & McNair Medical Institute, Houston, TX, USA
REFERENCES
- Shanik MH et al. 2008 Diabetes Care 31 Suppl 2 S262–S268.
- Vafiadis P et al. 1996 Journal of Autoimmunity 9 397–403.
- Redondo MJ et al. 2018 Pediatric Diabetes 19 346–353.
- Oram RA et al. 2014 Diabetologia 57 187–191.
- Alemzadeh R et al. 1998 Journal of Clinical Endocrinology & Metabolism 83 1911–1915.
- Lustig RH et al. 2003 Journal of Clinical Endocrinology & Metabolism 88 2586–2592.
- Mehran AE et al. 2012 Cell Metabolism 16 723–737.
- Templeman NM et al. 2017 Cell Reports 20 451–463.
- Pedersen DJ et al. 2015 Molecular Metabolism 4 507–518.
- McCarthy MI 2017 Diabetologia 60 793–799.
- van Raalte DH & Verchere CB 2017 Diabetes, Obesity & Metabolism 19 1205–1213.
- ORIGIN Trial Investigators 2012 New England Journal of Medicine 367 319–328.
- Marso SP et al. 2016 New England Journal of Medicine 375 311–322.
- Zinman B et al. 2015 New England Journal of Medicine 373 2117–2128.
- Look ARG et al. 2013 New England Journal of Medicine 369 145–154.
- Lean ME et al. 2018 Lancet 391 541–551.
- Sutton EF et al. 2018 Cell Metabolism 27 1212–1221 e3.
- Sainsbury E et al. 2018 Diabetes Research & Clinical Practice 139 239–252.
- Hallberg SJ et al. 2018 Diabetes Therapy 9 583–612.
- Tay J et al. 2018 Diabetes, Obesity & Metabolism 20 858–871.
- Vafiadis P et al. 1997 Nature Genetics 15 289–292.
- Szabat M et al. 2016 Cell Metabolism 23 179–193.
- Sartori DJ et al. 2014 Molecular Endocrinology 28 28–39.
- Lamb MM et al. 2015 Diabetologia 58 2027–2034.
- Ferrara CT et al. 2017 Diabetes Care 40 698–701.
- Pugliese A 2017 Journal of Clinical Investigation 127 2881–2891.
- Brereton MF et al. 2014 Nature Communications 5 4639.
- El-Osta A et al. 2008 Journal of Experimental Medicine 205 2409–2417.
- Lennerz BS et al. 2018 Pediatrics 141 e20173349.



{kind=link}
{kind=link}