FIRST PUBLISHED IN ISSUE 77 (2005)
Polycystic ovary syndrome (PCOS) is the most common endocrine disorder in women of reproductive age, affecting 5–15% of the female population. PCOS has a huge and possibly insufficiently acknowledged impact on the nation’s health, as an early marker disease for cardiovascular risk.
At least half the women with PCOS are insulin-resistant and at an increased risk of developing the metabolic syndrome, including obesity, hyperlipidaemia, high blood pressure and type 2 diabetes. Furthermore, chronic anovulation due to PCOS is the most frequent cause of female infertility. Related treatment consumes the largest part of fertility treatment costs in the UK. Pregnancy in PCOS, once achieved, is not straightforward either, with a tenfold-increased risk of gestational diabetes and pregnancyrelated hypertension.
PCOS was first described in 1935 by Stein and Leventhal, who found bilateral polycystic ovaries in women with amenorrhoea at autopsy. Nowadays, luckily, we can catch these women earlier. Many of them initially present with mild obesity and oligomenorrhoea and hirsutism. Motivating them to comply with treatment may help to avoid the detrimental consequences of full-blown metabolic syndrome in later life. It is often useful to realise that these women may be genetically advantaged, as they would be the most likely to survive and even reproduce in times of extreme food restriction like poverty, hunger and war.
Though it is obvious to doctors that the term ‘syndrome’ implies that the underlying cause of disease is unclear, patients tend to interpret ‘polycystic ovary syndrome’ quite literally and anxiously look for treatment options to heal their ovaries. It is often difficult to explain the complexity of PCOS, that it is probably genetically determined and that it affects the whole endocrine–metabolic set-up. Patients therefore often wonder why dieting should get rid of cysts in their ovaries.
Most confusingly, the ultrasound appearance of polycystic ovaries is not necessarily a precondition for diagnosis. Two of the following are required for diagnosis of PCOS, according to the Rotterdam 2003 PCOS Consensus Workshop: (a) oligo- or amenorrhoea, (b) clinical or biochemical signs of hyperandrogenaemia, (c) polycystic appearance of ovaries at ultrasound (as defined by more than 12 cysts with a diameter of 2–9 mm).
Furthermore, polycystic ovaries are not necessarily indicative of PCOS. The work of Steve Franks and others tells us that polycystic ovaries at ultrasound are seen in a considerable number of women, without concurrent clinical evidence of the syndrome. Whether polycystic ovaries without PCOS represent a preclinical stage of the disease or an unrelated entity is a matter for debate.
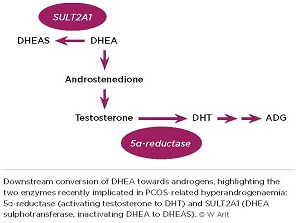
Importantly, endocrine researchers, like patients, tend to restrict their view of PCOS to the ovary, especially when considering the origin of hyperandrogenaemia, one of the syndrome’s main clinical features. In vitro studies have shown excess androgen production by theca cells isolated from ovaries in PCOS. This includes increased activity of 5α-reductase, resulting in increased conversion of testosterone to dihydrotestosterone (DHT), which binds the androgen receptor with tenfold higher affinity than testosterone.
However, whilst this seems to imply that the problem lies primarily in the ovary, earlier studies by Paul Stewart and colleagues provided indirect evidence that 5α-reductase activity may generally be enhanced in PCOS. They found that patients had increased urinary baseline excretion of 5α-reduced androgen and glucocorticoid metabolites. We have now revisited this concept using a dehydroepiandrosterone (DHEA) challenge test.
DHEA is a crucial precursor of human androgen synthesis, and we have previously shown that oral administration of DHEA leads to its efficient conversion towards androgens in women. Comparing women with PCOS and healthy controls, the oral DHEA challenge, preceded by dexamethasone suppression, was a tool to detect differences in downstream androgen generation.
While levels of androstenedione and testosterone following DHEA administration did not differ between groups, women with PCOS showed significantly higher generation of DHT and the DHT metabolite ADG, i.e. upregulation of all androgens downstream of 5α-reductase (see Figure).
This was further supported by urinary steroid excretion analysis, which revealed a significantly increased excretion of 5α-reduced androgen, glucocorticoid and mineralocorticoid metabolites following DHEA administration. A general increase in 5α-reductase activity will inevitably lead to enhanced androgen activation by conversion of testosterone to DHT in peripheral target cells of androgen action. This clearly highlights the importance of liver and other target tissues in a novel, ‘beyond the ovary’ perspective on hyperandrogenaemia in PCOS.
Our recent study on the interconversion of DHEA and its sulphate ester, DHEA sulphate (DHEAS), may have exposed another novel perspective.
Only desulphated DHEA is biologically active and can be converted towards androgens. It has previously been assumed that DHEA and DHEAS interconvert freely and continuously, DHEAS being activated to DHEA by steroid sulphatase, and DHEA inactivated to DHEAS by DHEA sulphotransferase (SULT2A1). So DHEAS is usually seen as a circulating storage pool for continuous DHEA regeneration.
However, while DHEA administration yields rapid generation of active androgens, we showed that administration of DHEAS did not lead to an increase in either DHEA or downstream androgens. This suggests that SULT2A1 activity, i.e. the inactivation of DHEA to DHEAS, appears to be the rate-limiting step regulating the DHEA–DHEAS equilibrium, determining DHEA bioavailability. These in vivo findings were supported by concurrent in vitro experiments, demonstrating ample generation of DHEAS from DHEA in cultured liver cells, but a complete lack of conversion of DHEAS to DHEA.
Serum DHEAS measurements are generally used as an estimate of adrenal androgen generation. However, from our recent findings, it seems highly likely that serum DHEAS may not appropriately reflect corresponding levels of desulphated, biologically active DHEA. Serum DHEA and DHEAS may be concordant in the physiological situation, but will be discordant in pathological conditions, in particular if SULT2A1 activity is impaired.
Hyperandrogenaemia of adrenal origin is usually excluded by measurement of serum DHEAS. A woman with normal DHEAS and increased androstenedione levels is generally considered to have hyperandrogenaemia of primarily ovarian origin. Most women with PCOS show this pattern, so seeming to justify the concentration of research into PCOS-related hyperandrogenaemia on mechanisms underlying ovarian androgen hypersecretion.
However, although serum DHEAS is normal, it may be that biologically active DHEA is pathologically increased, resulting in increased androstenedione through efficient downstream conversion of DHEA. Increased DHEA levels with concurrently low–normal DHEAS levels would suggest impairment of SULT2A1 activity. Preliminary findings in a large PCOS cohort, which we presented at BES 2005, revealed that a significant proportion of women with PCOS showed exactly that pattern.
One could argue that increased DHEA levels may result from enhanced ovarian CYP17 activity, the enzyme responsible for DHEA biosynthesis. However, we have shown that dexamethasone administration yields a similar and near complete suppression of serum DHEA in both healthy controls and women with PCOS, suggesting a primarily adrenal origin of circulating DHEA in both groups. This leaves SULT2A1 impairment as a potential novel mechanism underlying hyperandrogenaemia in PCOS.
These two mechanisms, enhanced 5α-reductase activity and putatively impaired SULT2A1 activity, illustrate the importance of steroidogenesis in understanding the pathophysiology of PCOS. A systemic view, rather than an ovarian spotlight, is likely to generate further insights into this fascinating disease.
Wiebke Arlt



{kind=link}