Pancreatic neuroendocrine tumours (PNETs) represent a spectrum of cancers that arise from the islet cells of the pancreas. PNETs are rare and reportedly account for 3–5% of all pancreatic tumours, with an incidence of 1–3 cases per 100,000 individuals per year. The incidence rate appears to be increasing. However, it is unclear whether this is a true increase or if it can be explained by improved detection on endoscopy and imaging.
Despite high profile cases, including Steve Jobs, Arethra Franklin and Irfan Khan, who have been diagnosed with and subsequently died from PNETs, there continues to be a lack of awareness and understanding of these tumours among the general public, and the medical and scientific communities. This may partly be explained by the relatively good prognosis and slow-growing nature of these tumours when compared with pancreatic ductal adenocarcinoma (PDAC), the most common type of pancreatic cancer.
However, a proportion of PNETs (e.g. poorly differentiated neuroendocrine carcinomas) are highly aggressive, with a median survival of <12 months, not dissimilar to that of PDAC. Furthermore, around 60% of patients present with metastatic PNETs, usually with liver metastasis, with a median survival of only 24 months.
Surgery is the mainstay of therapy for PNETs. However, this is often not curative due to advanced disease. Although currently available medical therapies have been shown to improve progression-free survival (surrogate marker for overall survival), many studies group PNETs with other types of NETs due to their rarity, making specific inferences regarding PNET outcomes challenging. Therefore, there is a need for more effective targeted treatments, based on an improved understanding of the molecular mechanisms that underpin the development of PNETs.
PNET GENETICS AND EPIGENETICS
Approximately 10–15% of PNETs occur as part of an inherited syndrome, such as multiple endocrine neoplasia type 1 (MEN1), von Hippel–Lindau disease, neurofibromatosis type 1 and tuberous sclerosis complex 1 and 2. Of these, MEN1 is the most common hereditary syndrome associated with an increased susceptibility to PNET development (approximately 10%).
However, the majority of PNETs are sporadic, with a low mutational burden. Nonetheless, large-scale whole genome and exome sequencing studies have shown that the mutations most commonly found in sporadic PNETs involve the genes MEN1, ATRX and DAXX, which are involved in the epigenetic mechanisms of histone modification and chromatin remodelling. Epigenetics (which literally means ‘above’ or ‘on top of ’ genetics) refers to changes in gene expression that are not caused by alterations in the underlying DNA sequence.
‘There is a need for more effective targeted treatments, based on an improved understanding of the molecular mechanisms that underpin the development of PNETs.’
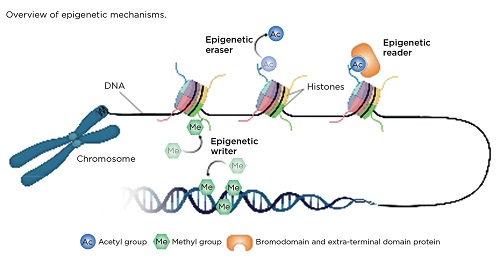
Enzymes responsible for carrying out epigenetic modifications are known as ‘writers’, while those that remove modifications are referred to as ‘erasers’. Epigenetic ‘readers’ have the ability to recognise and bind to specific epigenetic modifications and recruit additional proteins that regulate gene transcription (Figure). In cancer, these mechanisms may be dysregulated, resulting in either upregulation of oncogenes or downregulation of tumour suppressor genes. Unlike genetic mutations, epigenetic changes are reversible, therefore drugs that target epigenetic mechanisms may represent a promising therapy for the treatment of PNETs.
In October 2019, we formed the Oxford NETs research team, within Raj Thakker’s lab at the University of Oxford. The focus of our group is to investigate different aspects of epigenetics in NETs (including DNA methylation and histone modifications) as well as testing of novel epigenetic-targeting compounds.
THE DNA METHYLOME OF PNETs
DNA methylation is essential for normal development through its ability to silence/‘turn off’ specific genes. This process is required for cellular differentiation and homeostasis.
In DNA methylation, DNA methyltransferase (DNMT; an epigenetic writer) adds a methyl group to the DNA base cytosine (C) to form 5-methylcytosine (5mC), usually at the start of the gene sequence (promoter). This ‘mark’ can either be maintained or removed during cellular replication, or it can be actively removed by the epigenetic eraser ten-eleven-translocase (TET) to form 5-hydroxymethylcytosine (5hmC). In opposition to 5mC, 5hmC protects the promoter site and enables gene transcription.
Unbalanced DNMT and/or TET activity may lead to inappropriately methylated/‘switched off’ tumour suppressor genes or hydroxymethylated/‘switched on’ oncogenes resulting in uncontrolled cellular proliferation.
TARGETING HISTONE MODIFICATIONS IN PNETs
Histones, which package and condense DNA, can be epigenetically modified through the addition of methyl and/or acetyl marks to histone tails (read by epigenetic readers). This results in either an open/‘relaxed’ form (euchromatin), which enables gene transcription, or an inaccessible/‘closed’ form (heterochromatin).
The bromodomain and extra-terminal domain (BET) proteins are a family of epigenetic readers that bind to a specific type of histone modification (acetylation) and recruit other proteins that regulate gene expression. Dysfunction of the BET proteins has been linked to many cancers, through the upregulation of cancer-related genes, such as c-MYC.
Our group has previously shown that the BET inhibitor JQ1 decreases proliferation in PNET cell lines and increases apoptosis of PNETs that develop in mice. However, the poor half-life and bioavailability of JQ1 mean that it is unsuitable for clinical development. Novel BET inhibitors with improved selectivity and tolerability profiles have recently been developed. We are currently testing these and a panel of novel compounds (e.g. writers and erasers) in preclinical PNET models, to identify candidates that could be further developed for clinical use.
The variable efficacy and limited treatment options available for advanced PNETs presents a challenge for healthcare professionals, scientists and – most importantly – patients. In the current era of precision medicine, overcoming this challenge through an in-depth understanding of the epigenetic mechanisms involved in PNETs and the investigation of therapies that target these pathways may identify a novel treatment approach for these tumours.
KATHERINE A ENGLISH, OMAIR A SHARIQ AND KATE E LINES
Cancer Research UK Clinical Research Training Fellow, Clinical Research Fellow and Clarendon Scholar, and Oxford-Bristol-Myers-Squibb Research Fellow, Academic Endocrine Unit, University of Oxford, Oxford Centre for Diabetes, Endocrinology and Metabolism, Churchill Hospital, Oxford



{kind=link}