The parathyroid glands have a critical role in regulating serum calcium concentrations by secreting parathyroid hormone (PTH). Almost 30 years ago, the G protein-coupled receptor (GPCR) that allows parathyroid glands to detect serum calcium concentrations was cloned from bovine parathyroid tissue and named the calcium-sensing receptor (CaSR). Studies since then have helped us understand that calcium binding to the extracellular surface of the CaSR triggers receptor conformational changes, activation of associated G proteins and their downstream signalling pathways and, ultimately, suppression of PTH secretion
The CaSR is able to couple to all four G protein family subtypes, but predominantly activates two families, Gq/11 and Gi/o, to stimulate intracellular calcium mobilisation and MAPK pathways, and reduce cAMP production. Studies in the past 10 years have, however, revealed complexities within the CaSR signalling system and expanded our understanding of calcium homeostasis (Figure).

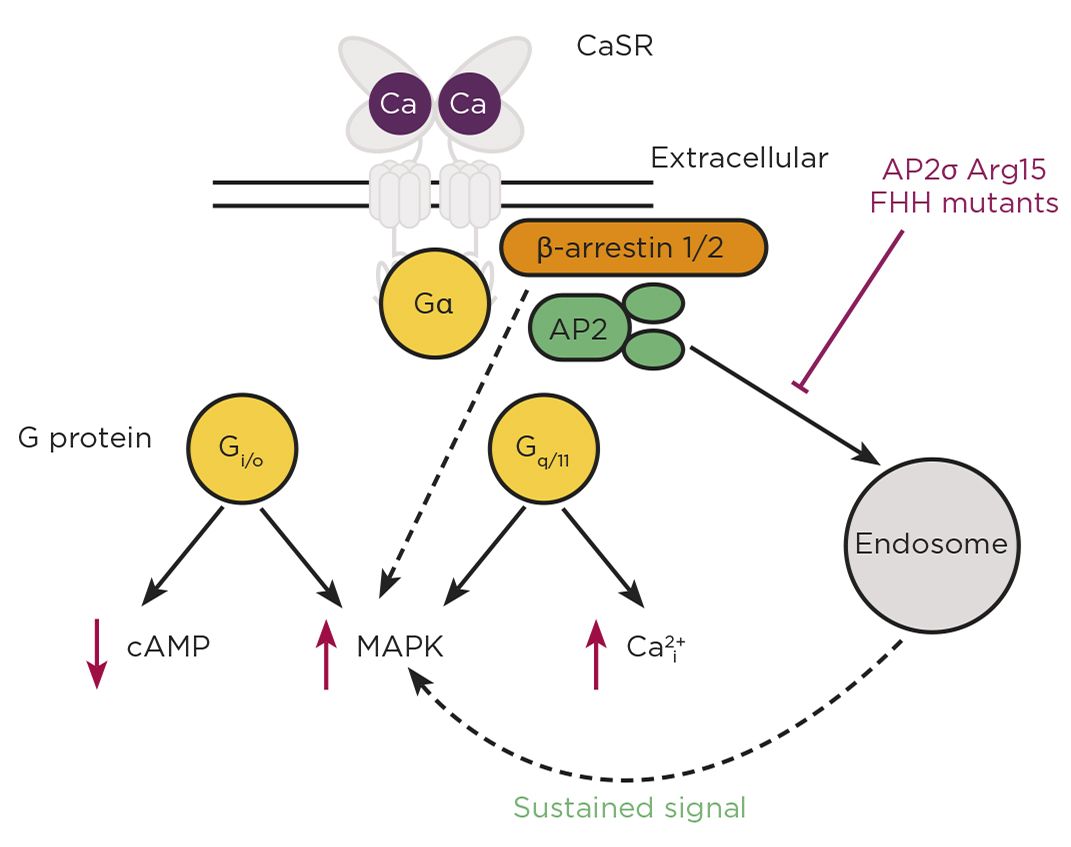
The CaSR can activate G protein-signalling pathways downstream of the G proteins Gi/o and Gq/11, leading to cAMP reductions, and increases in calcium mobilisation and MAPK signalling. Mutations in the CaSR, Gα11 and AP2σ proteins cause disorders of calcium homeostasis. CaSR can signal via a G protein-independent pathway involving β-arrestin and can signal from within the cell following endocytosis (sustained signal). FHH, familial hypocalciuric hypercalcaemia.
CaSR MUTATIONS DISRUPT CALCIUM HOMEOSTASIS
The publication of the CaSR gene sequence led to a rapid succession of manuscripts describing mutations in the receptor associated with disorders of calcium homeostasis. It is now understood that inactivating mutations cause familial hypocalciuric hypercalcaemia (FHH) and, rarely, neonatal severe hyperparathyroidism, which can be fatal if untreated.1,2 In contrast, activating CaSR mutations cause autosomal dominant hypocalcaemia (ADH). Although benign in many cases, CaSR mutations can cause symptoms in some patients including muscle cramps, kidney stones, chondrocalcinosis and seizures.1,2
'The investigation of CaSR mutations in vitro has been important in confirming their pathogenicity, but has also revealed individual residues that play critical roles in CaSR receptor activation.'
The investigation of CaSR mutations in vitro has been important in confirming their pathogenicity, but has also revealed individual residues that play critical roles in CaSR receptor activation. These include residues located in the receptor homodimer interface and transmembrane regions, at which both activating and inactivating mutations have been identified. These residues, termed ‘switch residues’, are hypothesised to act as molecular switches that undergo conformational changes on ligand binding, and their mutation facilitates receptor structures that preferentially signal via Ca2+or pERK pathways3 (i.e. they ‘bias’ signalling). Additionally, other CaSR mutations have been described that bias signalling towards G protein-independent β-arrestin pathways.4 The physiological effect of CaSR signalling by these specific pathways remains to be explored in detail, but could allow the design of CaSR-targeting compounds with fewer side effects.
MUTATIONS IN THE PATHWAY CAUSE HYPER-/HYPOCALCAEMIA
While CaSR mutations are the most common cause of FHH and ADH, a subset of patients have mutations in other proteins that function within the CaSR signalling and trafficking pathways.2 Both inactivating and activating mutations have been identified in the gene encoding Gα11, through which CaSR signals. These mutations cause a milder form of FHH, attributed to the ability of CaSR to compensate for loss of Gα11 signalling by coupling to other G proteins. This could explain why patients with Gα11 mutations do not present with a wider range of symptoms, despite numerous GPCRs with critical roles in the cardiovascular and central nervous systems utilising Gα11 signalling. Detailed studies of G protein signalling in the presence of mutant Gα11 are required to better understand this.
Perhaps most intriguingly, mutations in the σ-subunit of the adaptor protein-2 (AP2) also give rise to FHH. AP2 is a heterotetrameric protein that plays a fundamental role in clathrin-mediated endocytosis, a process required by cells for nutrient uptake and internalisation of transmembrane proteins. AP2σ is the smallest subunit of the AP2 complex, and mutations in FHH patients affect a single residue, Arg15. Previous studies show that this residue is important for binding to internalisation motifs within transmembrane proteins. We therefore hypothesised that FHH mutations in Arg15 must affect binding of AP2σ to CaSR, and thus affect CaSR internalisation. Indeed, studies of CaSR trafficking using total internal reflection fluorescence microscopy demonstrated an impairment of CaSR endocytosis in cells expressing FHH-associated AP2σ mutations, and consequently increased CaSR cell surface expression.5 However, AP2σ mutations reduce CaSR-mediated signalling. At first, this appeared paradoxical, as more receptor at the cell surface should equate to more signalling by the receptor. So, how are the AP2σ mutations impairing CaSR signalling?
To uncover the molecular mechanisms, we turned to other GPCRs as a guide. Some GPCRs, including thyrotrophin and PTH receptors, can continue to signal once internalised.6 We hypothesised that the CaSR may also have long-lasting cytoplasmic signals, and that depletion of internalised CaSR by the AP2σ mutations would reduce signalling from this source. Using a combination of imaging techniques, signalling assays and inhibitors of endocytosis, we showed that CaSR can signal from an internal location that is likely to be endosomal.5 Furthermore, this internalisation-dependent pathway involves only Gq/11 signalling and, therefore, these studies demonstrate how a single GPCR can resolve pleiotropic signals by spatially directing G protein selectivity.
REMAINING QUESTIONS
Elucidation of these novel CaSR signalling pathways has uncovered new mechanisms by which the receptor signals. However, they have generated important outstanding questions, such as:
- what are the physiological functions of CaSR endosomal signalling?
- does endosomal signalling take place in all CaSR-expressing cells?
Further research focused on answering these questions may help facilitate the development of targeted therapies, to activate sustained endosomal signalling in a biased manner. This could provide novel pharmacological treatments for a range of hyper- and hypocalcaemic disorders.
CAROLINE GORVIN
Institute of Metabolism and Systems Research, University of Birmingham
REFERENCES
- Gorvin CM 2019 Journal of Molecular Endocrinology 63 R1–R16.
- Hannan FM et al. 2016 Journal of Molecular Endocrinology 57 R127–R142.
- Gorvin CM et al. 2018 Human Molecular Genetics 27 3720–3733.
- Gorvin CM et al. 2018 Science Signaling 11 eaan3714.
- Gorvin CM et al. 2018 Cell Reports 22 1054–1066.
- Hanyaloglu AC 2018 International Review of Cell & Molecular Biology 339 93–131.




{kind=link}