Takotsubo syndrome (TTS), also known as ‘broken heart syndrome’, is an increasingly recognised condition which represents a large overlap between endocrinology and cardiology. It is a severe, but reversible, acute heart failure that occurs following a catecholamine surge.
TTS patients present similarly to those with myocardial infarction (MI), with chest pain and ST-segment elevation on electrocardiogram. Critically, they are distinguished by imaging, with left ventricular apical hypokinesia with basal hypercontractility occurring in the absence of culprit coronary artery disease. This characteristic pattern led to the naming of TTS, with ‘tako-tsubo’ translating to ‘octopus pot’ in Japanese, which its morphology resembles.
TTS predominantly affects postmenopausal women, typically occurs following extreme physical or emotional stress,1 and is thought to represent 5–6% of female patients presenting with suspected ST-elevation MI, with similar prognosis.2 The profound contractile dysfunction in TTS causes serious complications, including cardiogenic shock, thrombi formation, left ventricular rupture, pulmonary oedema and arrhythmia. There is no evidence-based treatment for the acute or chronic management of TTS, therefore a greater understanding TTS pathogenesis is important.
Whilst the association of TTS with catecholamines is well evidenced, there is no single established pathophysiological mechanism. Endothelial dysfunction, direct catecholaminergic myocardial stunning, oestrogen deprivation and brain–heart interaction have all been identified as important in TTS.
CATECHOLAMINERGIC MYOCARDIAL STUNNING
There is good evidence that catecholamines play a causative role in TTS. TTS may occur secondary to medical conditions with elevated catecholamine levels, such as phaeochromocytoma, acute subarachnoid haemorrhage and acute thyrotoxicosis, and, iatrogenically, following dobutamine stress echocardiography or adrenaline administration. Further, TTS is robustly induced in rodent and primate models with adrenaline.3,4
Whilst the β1-adrenergic receptor (β1AR) signals via the stimulatory Gαs pathway, the pleiotropic β2AR can signal via Gαs or the inhibitory Gαi. This limits the proarrhythmic and proapoptotic effects of Gαs activity and shifts receptor coupling to Gαi by a process known as stimulus trafficking or biased agonism. Consequently, β2AR-Gαi is directly cardiodepressive. βARs have the highest density in the ventricular apex,5 and apical cardiomyocytes have significantly larger overall βAR and β2AR responses than basal cardiomyocytes.3 This results in circulating adrenaline more profoundly affecting the apex of the ventricle in extreme stress.
β2AR-Gαi negative inotrope effects can be reversed by inhibition with pertussis toxin,3 although this also increases mortality in preclinical models. Endomyocardial biopsy studies from patients with TTS show activation of downstream β2AR-Gαi prosurvival pathways. Indeed, increased levels of G protein-coupled receptor kinase and β-arrestin have been identified in TTS patients,6 and these proteins are required for β2AR-Gαi stimulus trafficking to occur.
OESTROGEN DEPRIVATION
Given the increased incidence of TTS in postmenopausal women, the involvement of oestrogen withdrawal has, logically, been proposed. Oestrogen is cardioprotective via a multitude of mechanisms, including limiting sympathetic stimulation and activating β2AR-Gαi signalling pathways. Circulating adrenaline is lower in women than men at baseline, as are urinary cortisol, adrenaline and noradrenaline, which rise with age. Interestingly, these hormones are reduced by hormone replacement therapy (HRT), suggesting regulation by oestrogen. Adrenergic-dependent gene expression changes are observed in rodent models of TTS and prevented by oestrogen supplementation.7 The impact of HRT on TTS incidence and recurrence is not clear, and offers an interesting avenue for further investigation.

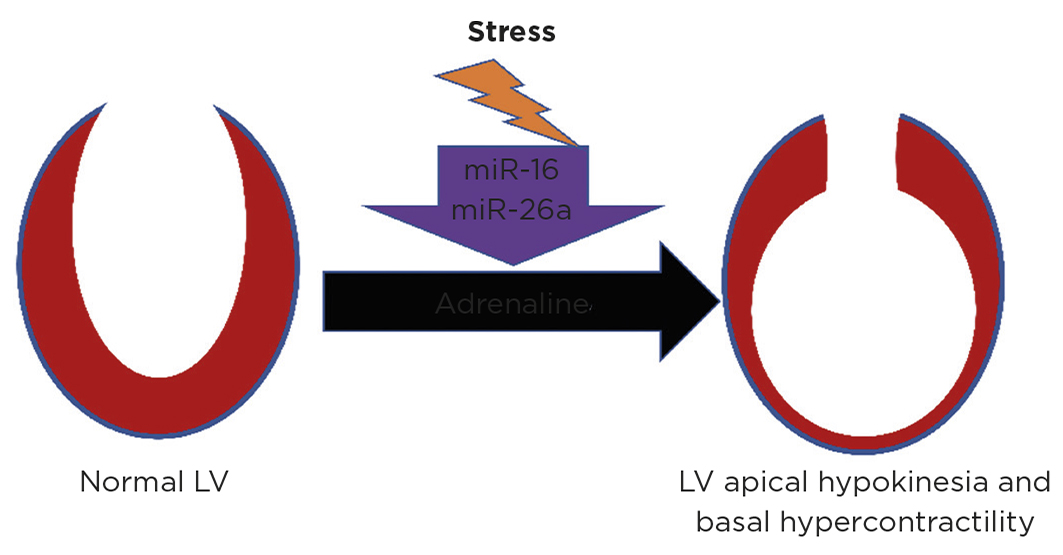
Figure. Prior stress may predispose to future TTS. Representation of the left ventricle (LV) showing TTS, with contractile dysfunction following adrenaline made more likely by prior neuropsychiatric stress. Reproduced under CC BY 4.0 Licence (http://creativecommons.org/licenses/by/4.0) from Couch et al.10 https://doi.org/10.1093/cvr/cvab210 ©2021 The Authors
BRAIN–HEART AXIS
Given the implied importance of the autonomic nervous system (ANS) in TTS, and the close association of TTS with neuropsychiatric disorders, there seems to be a clear interaction between the brain and heart in TTS.
Alterations within the insula, limbic system and ANS-specific signalling networks have been noted in patients with TTS.8 This suggests processing of emotions and subsequent ANS response to stress may be profoundly altered in TTS patients, and could explain the ‘gain’ in the hypothalamic-pituitary axis that has previously been suggested in TTS.1
MicroRNAs (miRNAs) have recently been identified that are linked to neuropsychiatric stress and can be used as biomarkers to distinguish TTS from ST-elevation MI and healthy controls.9 A recent preclinical study increased expression of TTS miRs (miR-16 and miR-26a) in vitro and in vivo. This predisposed to catecholamine-induced negative inotropism in the left ventricular apex and positive inotropism in the left ventricular base, and TTS generation in vivo at lower adrenaline concentrations.10 This raises the possibility of a pathophysiological link between pre-existing stress and TTS via these miRs, and that prior neuropsychiatric stress may prime the heart to be more vulnerable to future TTS generation in situations of high adrenergic stress (see Figure).10
LIAM S COUCH
King’s College London BHF Centre, The Rayne Institute, St Thomas’ Hospital, London
ALEXANDER R LYON
Department of Cardiology, Royal Brompton Hospital, and National Heart and Lung Institute, Imperial College London
SIAN E HARDING
National Heart and Lung Institute, Imperial College London
References
- Lyon AR et al. 2016 European Journal of Heart Failure 18 8–27.
- Redfors B et al. 2015 International Journal of Cardiology 185 282–289.
- Paur H et al. 2012 Circulation 126 697–706.
- Izumi Y et al. 2009 Hypertension Research 32 339–346.
- Lyon AR et al. 2008 Nature Clinical Practice Cardiovascular Medicine 5 22–29.
- Nakano T et al. 2018 Scientific Reports 8 12731.
- Ueyama T et al. 2007 Circulation Journal 71 565–573.
- Hiestand T et al. 2018 Journal of the American College of Cardiology 71 809–812.
- Jaguszewski M et al. 2014 European Heart Journal 35 999–1006.
- Couch LS et al. 2021 Cardiovascular Research doi: 10.1093/cvr/cvab210.



{kind=link}