Triglycerides form the major component of dietary fat. They are hydrolysed by lingual, gastric, pancreatic and intestinal lipases into free fatty acids (FFA) and glycerol, which are re-esterified to triglycerides in enterocytes and packaged into the transport cargo, chylomicrons. Chylomicrons are hydrolysed by lipoprotein lipase (LPL), which is expressed at high concentration on the capillary surfaces of muscle and adipose tissue.
TRIGLYCERIDES AND SEVERE HYPERTRIGLYCERIDAEMIA
Definition of severity of hypertriglyceridaemia varies amongst different guidelines. However, normal levels of fasting triglycerides have consistently been described as <1.7mmol/l. The 2018 American Heart Association/American College of Cardiology Clinical Practice Guidelines and US National Cholesterol Education Programme Adult Treatment Panel III Guidelines have defined severe hypertriglyceridaemia as >5.6mmol/l, while the Endocrine Society and the European Atherosclerosis Society/European Society of Cardiology have defined this as >10.0mmol/l.1–4
Severe hypertriglyceridaemia is rare and is associated with genetic polymorphism that is often exacerbated by environmental factors. There are more than 300 independent gene loci that can affect plasma triglyceride levels with variable effect size. A single, extremely rare variant with a large effect size can lead to severe hypertriglyceridaemia (e.g. monogenic familial chylomicronaemia syndrome; FCS). An increase in mutational load of common variants with small effect size, compounded by environment factors, can cumulatively increase triglyceride levels (multifactorial chylomicronaemia syndrome; MCS).5,6
WHAT IS CHYLOMICRONAEMIA SYNDROME?
Chylomicronaemia is persistence of chylomicron particles in the circulation after a fasting period of 12–14 hours (they are normally cleared in 3–4 hours after a meal). This is associated with severe hypertriglyceridaemia (>10.0mmol/l). Chylomicronaemia syndrome is the presence of clinical abnormalities, such as eruptive xanthomas, lipaemia retinalis, recurrent abdominal pain, acute pancreatitis, hepatosplenomegaly, and neuropsychiatric or cognitive complications.
Monogenic causes of chylomicronaemia are extremely rare, with a population prevalence of 1–2 per million.7 The disease presents in childhood or early adolescence at the latest, and is characterised by severe hypertriglyceridaemia and recurrent episodes of pancreatitis, and is resistant to treatment. These are autosomal recessive disorders and characterised by a marked reduction in LPL activity, either due to mutation in the LPL gene or genes encoding proteins responsible for maturation, stabilisation, transport, anchoring and activation of LPL.
Most cases of chylomicronaemia are polygenic, due to the clustering of multiple genetic variants. An aggravating factor is usually required to augment the metabolic defect to reduce the clearance of chylomicrons. When enough of these variants are cumulatively inherited, they create a tendency to develop chylomicronaemia.5 In contrast to FCS, MCS has a population prevalence of 1:600, a milder phenotype and a relatively low risk of acute pancreatitis. It is generally more responsive to treatment or removal of secondary factors and lipid-lowering medications.
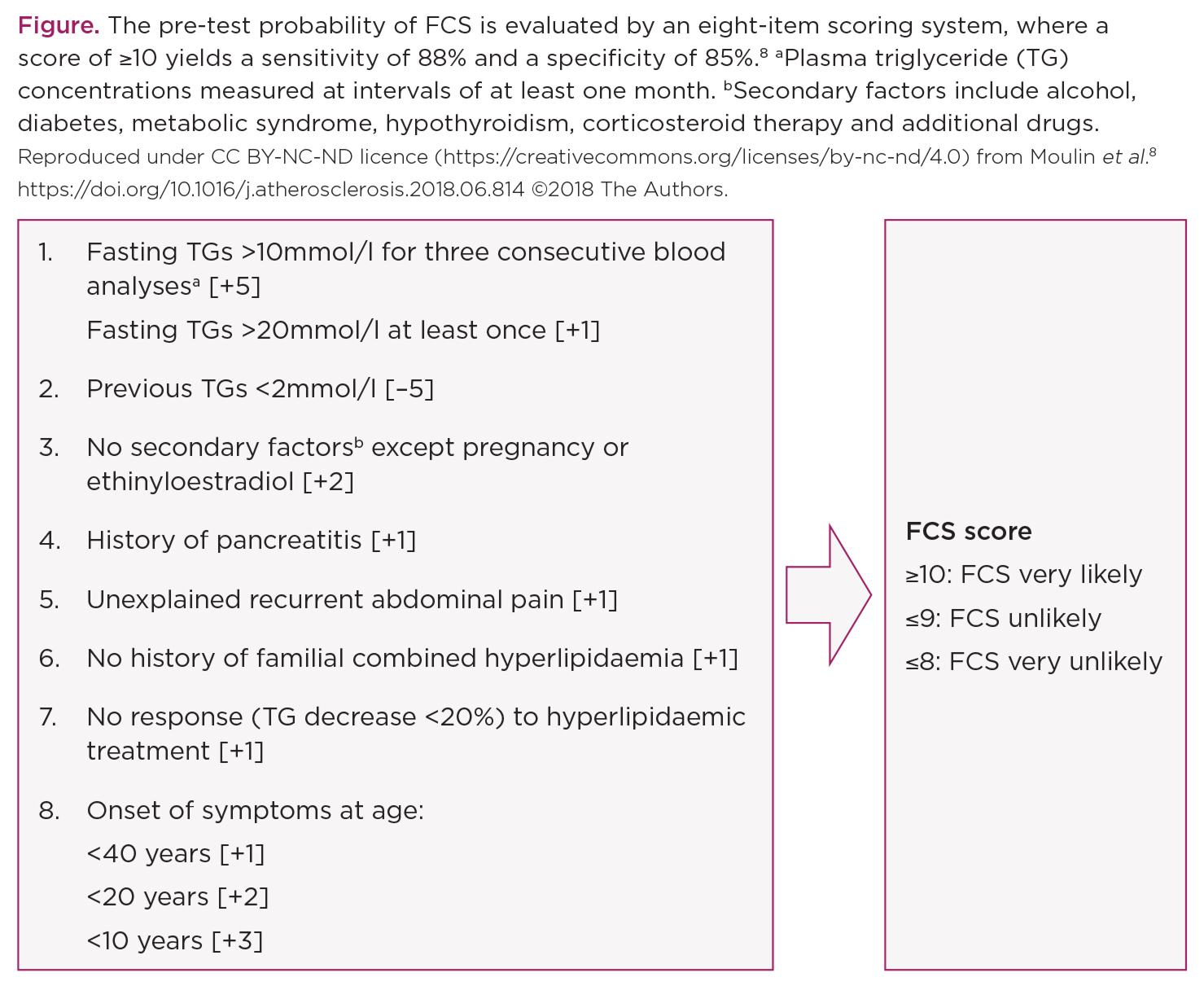
Metabolic phenotypes of FCS and MCS largely overlap, particularly during the periods of decompensation. The diagnosis of FCS is delayed in most patients, who suffer multiple attacks of acute pancreatitis before a diagnosis of FCS is made. While the gold standard to diagnose FCS remains genetic testing, the pre-test probability of FCS is evaluated by an eight-item scoring system, where a score of ≥10 yields a sensitivity of 88% and a specificity of 85% (Figure).8
ACUTE PANCREATITIS AND SEVERE HYPERTRIGLYCERIDAEMIA
Acute pancreatitis is the most serious complication of severe hypertriglyceridaemia, and its prevalence increases sharply if the level of triglycerides is >20mmol/l. Hypertriglyceridaemia constitutes only 5% of cases of acute pancreatitis, but it is associated with a more severe disease course, persistent organ failure, higher recurrence rate, greater length of hospital and intensive care unit stay and increased severity of complications and mortality when compared with other causes of acute pancreatitis.9
Although there is no correlation between severity of acute pancreatitis and severity of hypertriglyceridaemia, the disease course, recurrence and complications tend to be higher in FCS when compared with MCS.
There are no universally accepted guidelines for the management of hypertriglyceridaemia-induced pancreatitis and no realistic goals have been established. The probability of developing persistent organ failure can be reduced by rapidly reducing the triglyceride levels to <5.6mmol/l within the first 48 hours. This effect is time-sensitive, i.e. the earlier the goal is achieved the lower the likelihood of developing persistent organ failure.10
THERAPEUTIC OPTIONS IN ACUTE PANCREATITIS
Half of the mortality from acute pancreatitis happens within two weeks from the onset of symptoms. Bowel rest, intravenous hydration, pain management, restricted oral intake, low fat diet (<20g/day) and avoidance of oil-based medication (e.g. propofol) remain the key therapeutic interventions that should be employed as soon as the diagnosis is suspected.
‘There are more than 300 independent gene loci that can affect plasma triglyceride levels with variable effect size.’
Intravenous insulin infusion reduces triglyceride levels by up to 75%, by upregulating LPL activity. This may reduce the severity of pancreatitis, hasten the recovery and reduce the hospital stay. Intravenous heparin infusion reduces triglyceride levels early but, due to the risk of rebound hypertriglyceridaemia, worsening lipotoxicity from FFA and pancreatic haemorrhage, it is not recommended.
Lipoprotein and plasma apheresis not only remove chylomicrons and triglycerides rapidly from the circulation, they also remove proinflammatory cytokines to downregulate the inflammatory process in hypertriglyceridaemia-induced pancreatitis.11 Despite that, it has failed to demonstrate a reduction in morbidity or mortality, and is recommended only in individual cases who fail to improve despite conservative treatment, under specialist advice.
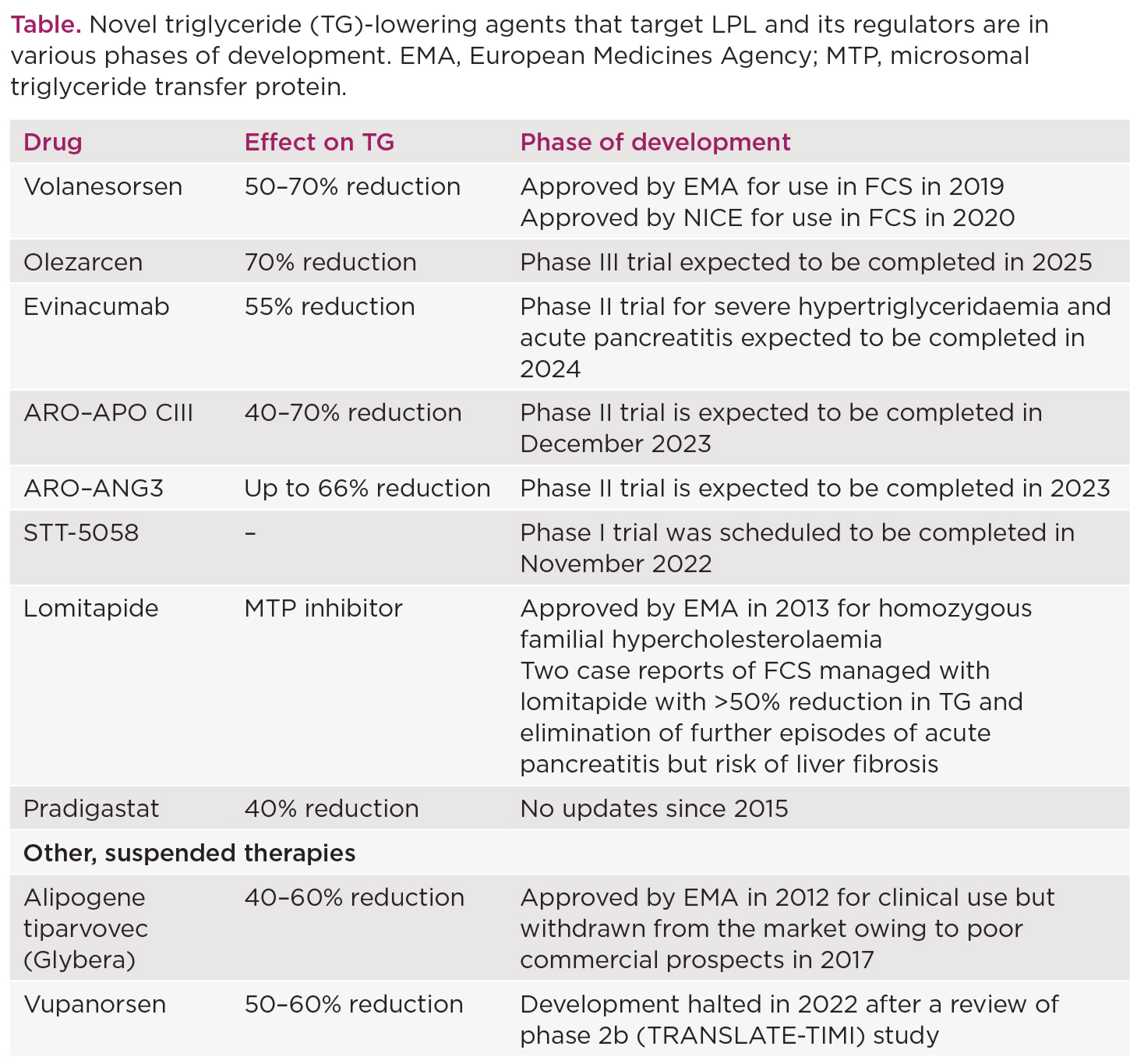
Of the pharmacological agents that are currently available, the lipid-lowering efficacy of fibrates, niacin and omega-3 fatty acids in chylomicronaemia syndromes is modest at best, with no effect on triglyceride levels in FCS. Several novel triglyceride-lowering agents that target LPL and its regulators are in various phases of development (Table). Volanesorsen was approved by NICE in 2020 as a therapeutic option for individuals with genetically confirmed FCS who are at risk of pancreatitis. It reduces serum triglyceride levels by 70–80% and the risk of recurrent pancreatitis. Thrombocytopenia remains a predominant concern with volanesorsen, requiring regular monitoring every one to two weeks. It is not advisable to start treatment if the platelet count is <140x109/l. No other drug has yet been approved for standard clinical use.
Chylomicronaemia syndromes pose a major clinical burden with potential to develop life-threatening pancreatitis. At present, low fat or fat-free diet and lifestyle modifications continue to be the cornerstone in the management of these syndromes, but are often difficult to adhere to. Novel therapeutic targets (Table) may offer solutions for their treatment and the prevention of associated complications.
Bilal Bashir and Handrean Soran
Centre of Diabetes, Endocrinology and Metabolism, Peter Mount Building, Manchester University NHS Foundation Trust; Faculty of Biology, Medicine and Health, University of Manchester; and Cardiovascular Trials Team, Manchester University NHS Foundation Trust
REFERENCES
1. Catapano AL et al. 2016 European Heart Journal 37 2999–3058.
2. Berglund L et al. 2012 Journal of Clinical Endocrinology & Metabolism 97 2969–2989.
3. Grundy SM et al. 2019 Circulation 139 e1082–e1143.
4. Multiple Authors 2002 Circulation 106 3143–3421.
5. Hegele RA et al. 2014 Lancet Diabetes & Endocrinology 2 655–666.
6. Carrasquilla GD et al. 2021 Current Atherosclerosis Reports 23 39.
7. Brahm AJ & Hegele RA 2015 Nature Reviews Endocrinology 11 352–362.
8. Moulin P et al. 2018 Atherosclerosis 275 265–272.
9. Wang Q et al. 2017 Journal of Clinical Gastroenterology 51 586–593.
10. Lu Z et al. 2020 Pancreas 49 105–110.
11. Garg R & Rustagi T 2018 BioMed Research International 2018 4721357.



{kind=link}
{kind=link}