Caroline Gorvin provides a synopsis of her Early Career Prize Lecture, covering work during her time as a Post-Doctoral Researcher at the Academic Endocrine Unit, University of Oxford.
Extracellular calcium (Ca2+e) is essential for the regulation of a multitude of biological functions, including mineralisation of bone matrix, neuromuscular excitability and blood coagulation, as well as for maintaining intracellular calcium (Ca2+i) stores that are required for functions ranging from muscle contraction to hormone secretion.1
The calcium-sensing receptor (CaSR) is a G protein-coupled receptor (GPCR) that plays a fundamental role in maintaining calcium homeostasis by sensing the Ca2+e level in the blood and regulating the synthesis of parathyroid hormone (PTH) to control this.2
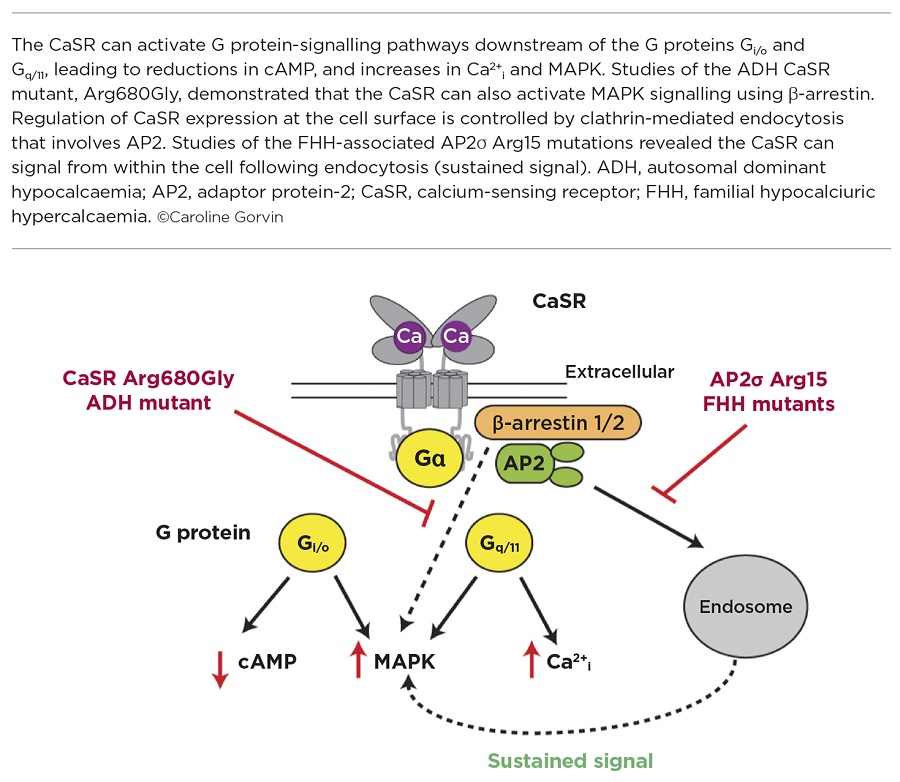
Ca2+e binds directly to the CaSR, which leads to receptor conformational changes that mediate GDP-to-GTP exchange at associated G proteins, and initiate downstream signalling pathways. The CaSR is able to couple to a variety of G proteins, but predominantly to the Gq/11 and Gi/o families, which activate signalling including initiating Ca2+i mobilisation from the endoplasmic reticulum, activating mitogen-activated protein kinase (MAPK) pathways and reducing cyclic AMP (cAMP) production.2
STUDYING CaSR MUTATIONS
Much of what has been learnt about the CaSR comes from studies of patients with mutations in the receptor. In the majority of cases, patients with heterozygous loss-of-function mutations of the CaSR have familial hypocalciuric hypercalcaemia (FHH), while patients with heterozygous gain-of-function mutations have autosomal dominant hypocalcaemia (ADH). Although benign in most cases, CaSR mutations can cause muscle cramps, kidney stones, chondrocalcinosis and seizures in some patients. Characterisation of the effect of these disease-causing mutations in vitro has demonstrated that they may influence CaSR signalling responses in a biased manner, with some FHH mutations switching the wild type CaSR from preferentially coupling to Ca2+i to a mutant receptor that signals predominantly by MAPK, or equally via the Ca2+i and MAPK pathways. In contrast, many ADH mutations cause a signalling bias by coupling more strongly to Ca2+i.3 However, the molecular mechanisms that mediate this signalling bias are often unclear.
THE Arg680Gly MUTATION
In our studies, we identified an ADH-associated mutation which affects the Arg680 residue of the CaSR, which is located within the third transmembrane domain of the receptor.4 Using in vitro analyses, it was shown that, in contrast to other ADH mutations, Ca2+i signalling is not affected by the Arg680Gly mutation, and that it instead enhances MAPK signalling (Figure). This increased MAPK signalling did not arise from G protein-dependent pathways, investigated by blocking each G protein pathway using specific inhibitors, but involved a β-arrestin specific pathway. This β-arrestin-mediated pathway does not function at physiological concentrations of Ca2+e in wild type CaSR-expressing cells.
Using a homology model of the CaSR built with the structure of the related GPCR, mGluR1, and engineered mutants of the CaSR, it was demonstrated that the Arg680 residue forms a salt bridge with an adjacent residue on the second extracellular loop of the CaSR. This salt bridge maintains the CaSR in an inactive conformation, and the Arg680Gly ADH mutation disrupts this salt bridge, probably allowing greater flexibility between the cytoplasmic regions of the CaSR transmembrane domains, and facilitating β-arrestin binding, which acts as a scaffold for MAPK signalling components.
These studies have, therefore, demonstrated a novel signalling pathway, which may be exploited for the development of new treatments for patients with mutations in the CaSR, or other GPCRs.
MUTATIONS IN OTHER GENES
In addition to CaSR mutations, studies of hypercalcaemic patients have demonstrated that FHH may arise from mutations in other genes.
Interestingly, mutations in a subunit (the σ-subunit) of the adaptor protein-2 (AP2), which is ubiquitously expressed, also give rise to FHH.5 AP2 is a heterotetrameric protein that plays a fundamental role in clathrin-mediated endocytosis, a process which is required by cells for nutrient uptake and internalisation of transmembrane cell surface proteins, such as GPCRs. The AP2σ protein is the smallest subunit of the AP2 complex, and mutations identified in FHH patients affect a single residue, Arg15.
Previous studies have shown that the AP2σ Arg15 residue is important for binding to specific regions (dileucine motifs) of transmembrane proteins.6 It was therefore hypothesised that FHH mutations in Arg15 must affect binding of AP2σ to the CaSR, and thus affect CaSR internalisation. Indeed, studies of CaSR trafficking using total internal reflection fluorescence microscopy demonstrated an impairment of CaSR endocytosis in cells expressing FHH-associated AP2σ mutations, and consequently increased CaSR cell surface expression.7 However, AP2σ mutations reduce CaSR-mediated signalling by both Gq/11- and Gi/o-mediated pathways. This at first appeared paradoxical, as more receptor at the cell surface should equate to more signalling by the receptor. So, how are the AP2σ mutations impairing CaSR signalling?
ELUCIDATING THE MECHANISM
To uncover the molecular mechanisms, we turned to other GPCRs as a guide. Some GPCRs, including the receptors for thyrotrophin, PTH and luteinising hormone, can continue to signal once internalised.8 We hypothesised that the CaSR may also have long-lasting signals from within the cytoplasm, and that depletion of internalised CaSR by the FHH-associated AP2σ mutations would reduce signalling from this source.
Using a combination of imaging techniques, signalling assays and inhibitors of endocytosis, it was demonstrated that the CaSR can signal from an internal location that is likely to be endosomal (Figure).7 Furthermore, this internalisation-dependent pathway involves only Gq/11 signalling and therefore these studies have shown how a single GPCR can resolve pleiotropic signals by spatially directing G protein selectivity.
Elucidation of these novel CaSR signalling pathways has uncovered new mechanisms by which the receptor signals, and may help facilitate the development of targeted therapies to activate β-arrestin-mediated or sustained endosomal signalling in a biased manner.
Caroline Gorvin, Senior Research Fellow, Institute of Metabolism and Systems Research, University of Birmingham
REFERENCES
- Brown EM 1991 Physiological Reviews 71 371–411.
- Conigrave AD & Ward DT 2013 Best Practice & Research: Clinical Endocrinology & Metabolism 27 315–331.
- Leach K et al. 2012 Endocrinology 153 4304–4316.
- Gorvin CM et al. 2018 Sci Signal In Press.
- Nesbit MA et al. 2013 Nature Genetics 45 93–97.
- Kelly BT et al. 2008 Nature 456 976–979.
- Gorvin CM et al. 2018 Cell Reports 22 1054–1066.
- Sposini S & Hanyaloglu AC 2017 Biochemical Pharmacology 143 1–9.




{kind=link}