While we often associate the term ‘stress’ with anxiety, the term really encompasses any challenge that has potential to disrupt homeostasis. This includes physical stressors like infection or injury. The physiological requirements in response to these diverse challenges are all mediated – at least in part – by the glucocorticoid cortisol.
PHYSIOLOGICAL EFFECTS
Glucocorticoids are cholesterol-derived steroid hormones synthesised and secreted by the adrenal gland. They are anti-inflammatory in all tissues, and control metabolism in muscle, fat, liver and bone. Glucocorticoids also affect vascular tone, and in the brain influence mood, behaviour and sleep‒wakefulness cycles.
Glucocorticoid excess (due to pathology such as Cushing’s syndrome or prescribed synthetic glucocorticoids) can cause immunosuppression, muscle atrophy, central adiposity, hepatosteatosis, osteoporosis, insulin resistance, hypertension, depression and insomnia.
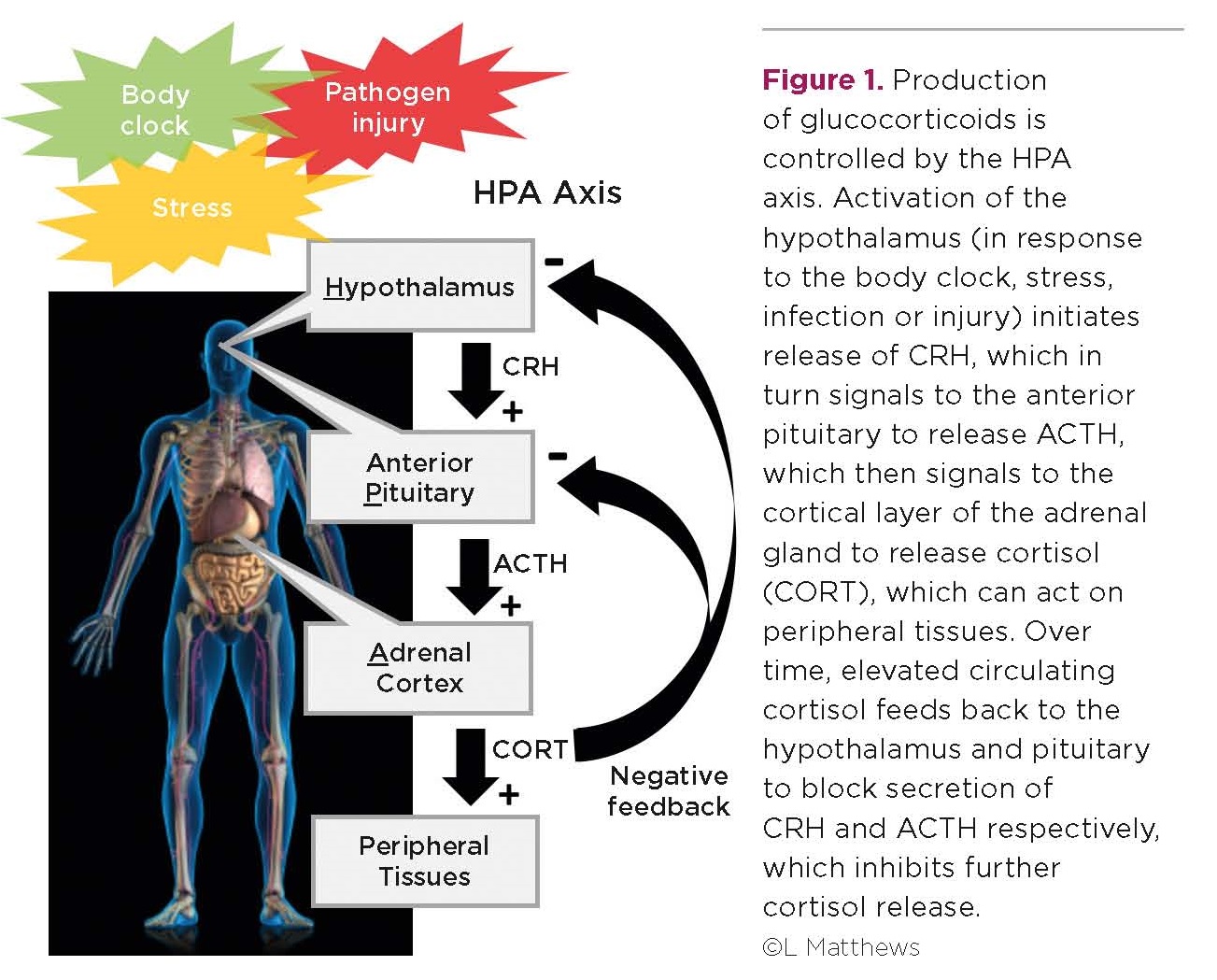
Elevated glucocorticoids can therefore be damaging, and so their production is under the strict control of the hypothalamic-pituitary-adrenal (HPA) axis (Figure 1). Activation of the hypothalamus initiates the release of corticotrophin-releasing hormone (CRH), which in turn signals to the anterior pituitary to release adrenocorticotrophin (ACTH). This then signals to the cortical layer of the adrenal gland to release glucocorticoids, which can act on peripheral tissues.
Over time, elevated circulating glucocorticoids feed back to the hypothalamus and pituitary to block further release of CRH and ACTH respectively, which inhibits glucocorticoid release. As a consequence, glucocorticoids are secreted in a pulsatile manner, and display a diurnal rhythm where circulating hormone levels peak at the onset of waking.
MAINTAINING BALANCE
In the context of maintaining normal homeostasis, the daily peak of glucocorticoid is very important. The transient peak increases vascular tone and alertness, mobilises energy and has a priming effect on the immune system. Essentially, your morning dose of cortisol prepares you to face the potential challenges in your day.
Stresses encountered through the day can drive additional pulses of glucocorticoids from the adrenal which drive sustained, elevated glucocorticoid levels. This produces the same physiological response but with a greater magnitude. In a ‘fight or flight’ context for example, glucocorticoids increase vascular tone and alertness, mobilise energy (prepare you to run) and prime the immune system (prepare you for injury). These preparatory effects increase the likelihood that the stressor can be overcome, so that normal homeostasis can be quickly restored.
It seems paradoxical that a potent immunosuppressive hormone would prime the immune system, but facilitating activation of immune cells is beneficial. It increases the ability of the immune system to detect pathogens/injury and respond accordingly, and it facilitates resolution and repair, to restore balance. In the context of chronic infection or disease, however, the role of glucocorticoids changes, in order to prevent escalation and limit damage.
Glucocorticoids act directly on cells within the damaged tissue to block production of inflammatory signals (cytokines, chemokines) that attract immune cells. They act directly on the immune cells to inhibit their ability to infiltrate the tissue, and in some cell types induce their death. Glucocorticoids also promote activation of scavenger immune cells that help remove cell debris, promote repair and prevent lasting damage (such as fibrosis). At the cellular level, glucocorticoids are able to control metabolism, inflammation, adhesion, migration and survival, in a cell-specific and context-specific manner.
MECHANISM OF ACTION
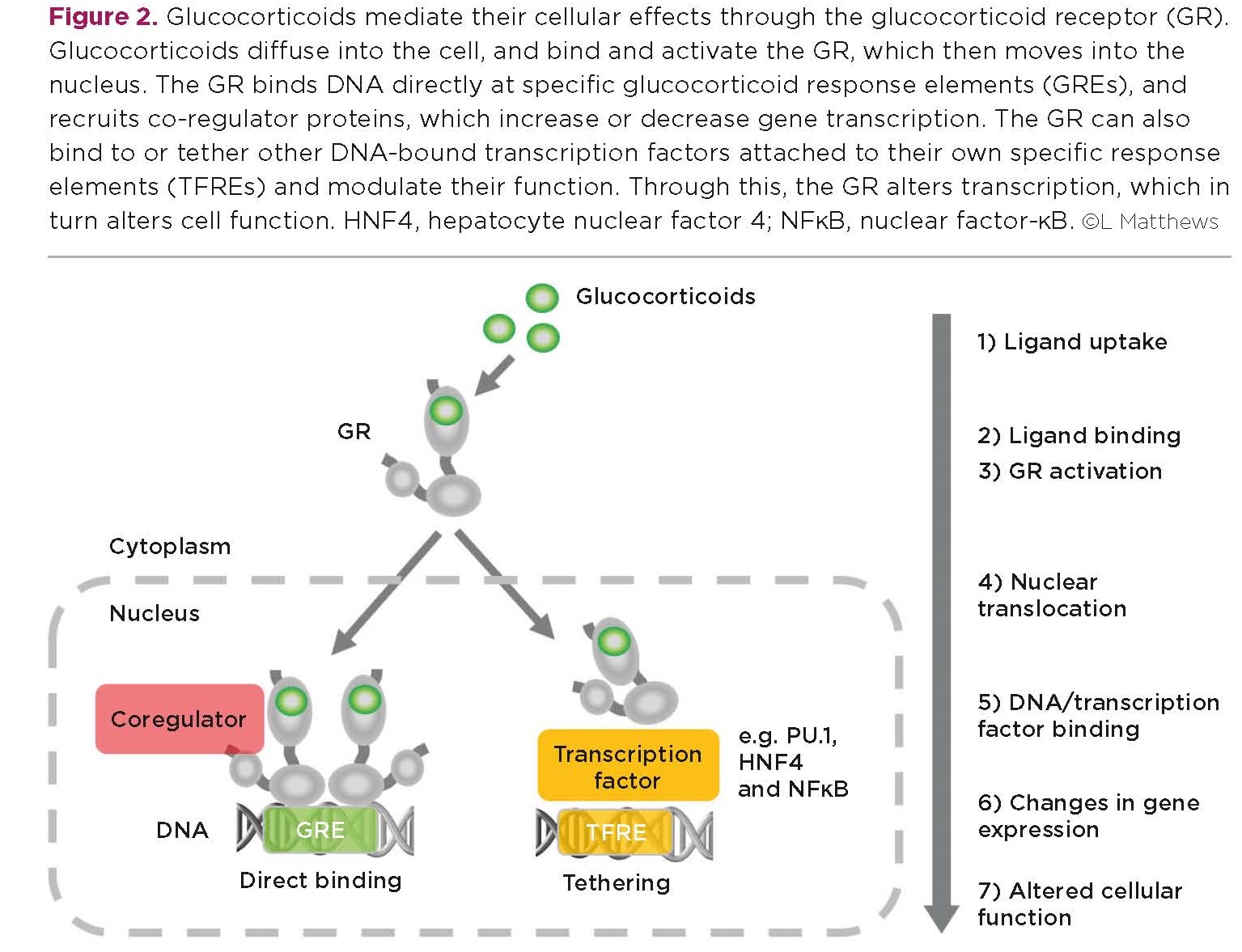
Glucocorticoids mediate their cellular effects through binding and activating the glucocorticoid receptor (GR), which is expressed in almost every tissue. Glucocorticoids diff use into the cell, and bind and activate the GR, which then moves into the nucleus.
The GR can bind DNA directly at specific glucocorticoid response elements (GREs), and then recruit coactivator or corepressor proteins to increase or decrease expression of target genes. The GR can also bind or tether itself to other DNA-bound transcription factors and modulate their ability to change gene expression (Figure 2).
The specific genes that the GR can control ‒ and therefore the cellular processes that glucocorticoids can regulate ‒ are critically determined by the ability of the GR to bind. This adds an additional layer of control, because gene accessibility and transcription factor expression and activity are cell-specific and dynamically regulated.
For example, liver-specific transcription factors such as HNF4 (hepatocyte nuclear factor 4) facilitate GR binding to (and regulation of) metabolic genes, whereas macrophage-specific transcription factors such as PU.1 recruit GR to genes important in immunity.
Glucocorticoid responses are therefore fine-tuned to consider context. This is how glucocorticoids can control metabolism in liver, activation of macrophages, and promote death of T-cells.
The GR is also recruited by the proinflammatory transcription factor NFκB (nuclear factor-κB). In contrast to HNF4 and PU.1, NFκB is expressed in every cell, but only binds DNA when activated in response to a pathogen or tissue damage. Consequently, the GR is only recruited to (and inhibits) proinflammatory genes if NFκB is activated. This explains why the GR (and glucocorticoids) is a potent inhibitor of inflammation only when required: i.e. when inflammation is already present.
Glucocorticoids (through the GR) are therefore perfectly adapted to integrate incoming signals from other pathways in order to respond appropriately to each specific challenge. We still have a long way to go in order to fully understand the whole spectrum of glucocorticoid action. However, we now have some insight into how, by adding a few additional points of control, glucocorticoids can co-ordinate diverse cellular effects to reach a common goal: the restoration of balance following stress.
Giorgio Caratti, Postdoctoral Fellow, University of Ulm, Germany
Pauline Pfänder, PhD Student, German Cancer Research Center (DKFZ), Germany
Laura Matthews, University Academic Fellow, University of Leeds



{kind=link}
{kind=link}