FIRST PUBLISHED IN ISSUE 116 (2015)
Congenital hypopituitarism (CH) is a rare disorder with a reported incidence ranging from 1 in 3,000 for isolated growth hormone (GH) deficiency to 1 in 10,000 for more complex disorders. It is associated with significant morbidity and, if undetected or inadequately treated, mortality.
Hypopituitarism is often associated with other congenital abnormalities such as eye and midline forebrain defects (septo-optic dysplasia, SOD), cervical vertebral abnormalities, cerebellar abnormalities and sensorineural hearing loss. Recent advances in our knowledge of the molecular mechanisms underlying these disorders have led to improved understanding of the conditions, and improved management at the bedside. Many of the genes identified in association with hypopituitarism encode transcription factors – these bind to DNA and either activate or repress transcription of downstream target genes.
THE MOLECULAR BASIS OF CH
Congenital hypopituitarism was believed to be a sporadic condition, the aetiology of which remained largely unknown. At one point, it was thought to be the result of birth trauma. In 1992, elucidation of the molecular basis underlying two dwarf mouse models, namely the Snell and Jackson dwarfs, led to the identification of the first gene associated with hypopituitarism in humans, PIT1 or, as it is now known, POU1F1.
POU1F1 AND PROP1 MUTATIONS
This was the first example of a candidate gene approach in the elucidation of the molecular basis of CH. Both recessive and dominant mutations have been identified in this gene encoding a pituitary transcription factor and, indeed, the most frequent mutation identified is the heterozygous missense substitution p.R271W, which can be transmitted in a dominant manner from one generation to the next.
The phenotype consists of GH and prolactin deficiencies, with variable thyrotrophin (TSH) deficiency, in keeping with a role for POU1F1 in the differentiation, proliferation and maintenance of somatotrophs, thyrotrophs and lactotrophs. Gonadotrophs and corticotrophs on the other hand are usually spared and so adrenocorticotrophin (ACTH) and gonadotrophin deficiencies are not associated with the phenotype.
A few years later, mutations in the pituitary-specific transcription factor PROP1 (or prophet of PIT1) were described in a further cohort of hypopituitary patients, following the identification of the molecular basis of the Ames dwarf mouse, which is the result of a recessive missense mutation in Prop1. The phenotype of the affected patients was largely one of GH, TSH and prolactin deficiencies, but with the addition of gonadotrophin deficiency. Initially, it was thought that the corticotrophs were spared, but it is now clear that ACTH deficiency is very much a part of the phenotype. However, the phenotype can evolve and ACTH deficiency in particular could be a late event.
A further feature of the phenotype is the transient enlargement of the pituitary gland, when the possibility of a tumour may be raised. However, the size of the pituitary can wax and wane before its eventual involution.
Generally, the phenotype of patients with PROP1 mutations is that of GH, TSH, prolactin, ACTH and gonadotrophin deficiencies, with a small anterior pituitary and a normally placed posterior pituitary. To date, PROP1 deficiency seems to be the commonest genetic cause of hypopituitarism, accounting for 50–100% of familial cases, although only around 10% of sporadic cases are due to PROP1 mutations.
HESX1 MUTATIONS
In 1998, the story became more complex with the identification of mutations in a transcriptional repressor gene called HESX1, initially in association with SOD, but later with GH deficiency and combined pituitary hormone deficiencies (CPHD) without midline or eye defects being added to the phenotypic spectrum.
SOD is a highly variable condition characterised by hypopituitarism, often including both anterior and posterior pituitary hormone deficiencies, in association with midline forebrain defects such as absence of the septum pellucidum and corpus callosum, and eye defects such as optic nerve hypoplasia and colobomas. Associated features include severe learning difficulties and autism. Only a small proportion (<1%) of SOD cases can be explained by HESX1 mutations; the majority remain unexplained, implying the existence of mutations in other genes that are still to be identified and/or epigenetic/environmental factors. One curiosity that is not yet understood is the increased incidence of SOD in younger mothers.
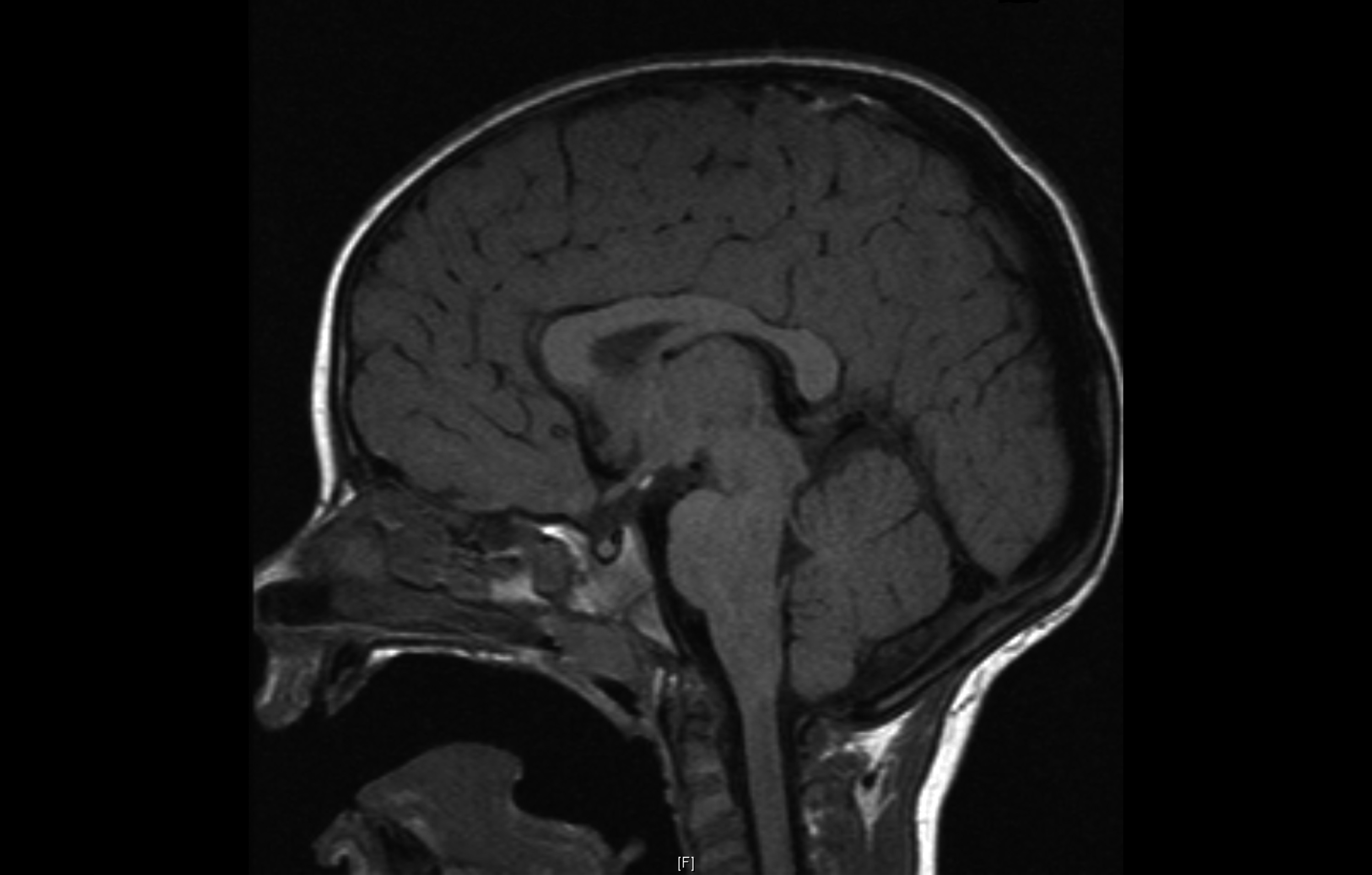
HESX1 mutations may be dominant or recessive, and have also been associated with pituitary aplasia. Additionally, the mutations may be variably penetrant; i.e. the parent can carry the same heterozygous mutation yet may not manifest the phenotype. The posterior pituitary is often placed ‘ectopically’ in the tuber cinereum or along the pituitary stalk – this may reflect a maldescent.

Ectopic posterior pituitary (PP) with anterior pituitary (AP) hypoplasia. Pituitary stalk (PS) is absent in this image. ©Mehul Dattani
LHX3 AND LHX4 MUTATIONS
In 2000, recessive mutations in LHX3 were described in patients with hypopituitarism and a short stiff neck with an abnormal cervical spine. Again, although it was initially thought that corticotrophs were spared, it is now clear that these patients will probably develop ACTH deficiency at some stage. Also, it has now been established that the majority of these patients exhibit a degree of sensorineural hearing impairment. The anterior pituitary may be small or occasionally enlarged, with the appearance of a microadenoma. The posterior pituitary is always eutopic, however. This is in contrast to mutations in the related gene LHX4, which can be associated with highly variable phenotypes including CPHD (mainly GH deficiency) associated with anterior pituitary hypoplasia and an ectopic posterior pituitary and cerebellar abnormalities on magnetic resonance imaging. The mutations are heterozygous and variably penetrant, with parental carriers often showing no phenotype.
OTHER MUTATIONS
More recently, duplications as well as loss of function mutations in the SRY-related gene SOX3 have been associated with X-linked hypopituitarism, classically GH deficiency but also panhypopituitarism, as well as variable learning difficulties. We have recently described a persistent craniopharyngeal canal in association with a SOX3 deletion. Heterozygous mutations, usually de novo, in the related gene SOX2 are associated with a complex phenotype consisting of learning difficulties, oesophageal atresia, severe eye abnormalities including anophthalmia, and hypogonadotrophic hypogonadism. SOX2 has been shown to be implicated in the proliferation and maintenance of stem cells in the pituitary, and continues to be expressed in the postnatal and adult pituitary in a small population of progenitors, the role of which remains to be established.
‘We can only identify a genetic basis in around 15% of cases, suggesting that other genes remain to be identified.’
Heterozygous mutations in OTX2 are also associated with severe eye abnormalities in association with hypopituitarism, classically GH deficiency, whereas variably penetrant mutations in GLI2 have been described in association with severe midline brain defects including holoprosencephaly in association with hypopituitarism and isolated CPHD without midline defects. The recent demonstration of a genetic overlap between hypopituitarism and Kallmann syndrome (PROKR2, FGFR1, FGF8, CHD7) has muddied the waters further.
Recent advances in genetic technology have led to the use of whole exome sequencing in the identification of novel candidate genes, and so this reflects a paradigm shift in approach. This approach has led to the identification of mutations in genes such as IGSF1 associated with X-linked central hypothyroidism, and ARNT2 associated with a severe recessive form of hypopituitarism, probably secondary to abnormal hypothalamic development, and vesico-ureteric reflux.
THE NEXT STEPS
‘Understanding the molecular basis of congenital hypopituitarism can have an impact on its clinical management.’
Much has been achieved in terms of our understanding of hypothalamopituitary development and the aetiology of CH. On the other hand, we can only identify a genetic basis in around 15% of CH cases, suggesting that other genes remain to be identified. The use of whole exome and potentially whole genome sequencing may lead to an improved understanding of the condition.
In particular, it is likely that, as observed with Kallmann syndrome, mutations in more than one gene may lead to the variable penetrance, and hence phenotypic expressivity, of hypopituitarism and SOD. Many other questions will need a more basic approach, for example the aetiology of the pituitary masses associated with PROP1 and SOX2 mutations. Close collaboration between basic scientists and clinicians will be required to further the field.
CLINICAL RELEVANCE OF MOLECULAR GENETICS IN CH MANAGEMENT
Understanding the molecular basis of CH can actually have an impact on its clinical management. For example, the identification of POU1F1 mutations would rule out the possibility of ACTH and gonadotrophin deficiencies, whereas the presence of PROP1 mutations might alert one to the probability of evolving ACTH deficiency, and encourage a more relaxed approach to the presence of a pituitary mass.
Although, at present, molecular analysis is offered in only a few centres on a research basis, it is likely that this addition to the diagnostic armamentarium for CH will be routine in the fullness of time.
Mehul Dattani, Currently Head of Paediatric Endocrinology at Great Ormond Street Hospital for Children (GOSH), University College London Hospitals (UCLH) and Institute of Child Health (ICH), London



{kind=link}