ANDROGEN RECEPTOR AND MICRORNAs
The androgen receptor (AR) is the critical driver of prostate cancer growth, both in early hormone-responsive disease and in advanced prostate cancer. Thus, the first-line treatment is androgen-deprivation therapy (ADT). However, resistance to this therapy usually develops after 2–3 years. This results in the emergence of lethal disease: so-called castration-resistant prostate cancer (CRPC). The mechanisms driving this progression remain largely unclear, although continued AR signalling despite depletion of circulating androgens is key, and may be partly driven by constitutively active AR variants (AR-Vs).
Notably, AR is the most microRNA (miR)-targeted oncogene in prostate cancer.1 MicroRNAs are small non-coding RNAs comprising around 22 nucleotides that bind to complementary sequences most often present within the 3’UTR of their target mRNAs, resulting in post-transcriptional regulation – usually, although not exclusively, negative. MicroRNAs can have oncogenic and/or tumour suppressive roles, with the potential to regulate a large number of genes/gene networks and thus have significant impact on disease outcome. Excitingly, they are also hormone-like, in that they are seemingly released (actively or passively) by tissues, circulate in the blood and so have the potential to act at distant sites, resulting in their description by the term ‘hormomiR’.2,3
MICRORNA TARGETING AND AR REGULATION
Mapping of miR:RNA interactions in prostate cancer cell lines has shown that, as well as being the most frequently targeted oncogene in prostate cancer, the AR is in the top 5% of all miR-regulated genes,1 supporting the disease-relevance of miR-mediated AR regulation. Post-transcriptional modulation of AR activity by miRs is via altering transcript stability, translational efficiency and regulatory networks.
Studies have identified a number of miRs that directly target the AR, including the miR-30 family members, miR-30c-5p and miR-30d-5p, which were shown to have significantly reduced expression in metastatic CRPC (mCRPC) compared with healthy control tissues.4 However, direct AR-modulatory miRs not only act to suppress the AR but can also increase AR activity, sometimes functioning as oncomiRs. Data from our laboratory have shown that miRs-346 and 361-3p, both of which directly bind the AR 3’UTR, increased not only the activity and expression levels of the wild type AR, but also that of AR-Vs, as well as promoting variant- driven prostate cancer cell proliferation. This upregulation could provide a mechanism by which the cancer cells maintain AR activity even under androgen-depleted conditions.5
Regulation of AR is not only achieved via the direct action of miRs, but also by a wide variety of protein factors, including co-repressors, co- activators, and pioneer and transcription factors. At least 200 different co-regulators are known to regulate the AR, many of which are themselves under miR control, thus constituting indirect miR regulation of AR activity. For example, the AR co-repressor SHP is repressed by oncogenic miR-141, which shows increased expression in prostate cancer, with the net result being increased AR activity.6 In contrast, AR activity is repressed indirectly by let-7c targeting of c-Myc, a transcription factor that positively regulates the AR gene.7
MicroRNAs are known to be dysregulated in CRPC when compared with hormone- responsive prostate cancer, with more decreased than increased at this later stage. CRPC-downregulated miRs often inhibit oncogenic processes such as proliferation, migration and invasion.
An example of one such is miR-145-3p, which functions as a tumour suppressor in prostate cancer by inhibiting cell cycle progression and survival through its targeting and downregulation of genes such as MELK, BUB1 and CDK1, all of which have been shown to be predictive for patient survival.8
ANDROGEN RECEPTOR VARIANTS
Expression of alternatively spliced forms of AR, known as AR-Vs, is an important mechanism of ADT resistance. These variants lack the ligand-binding domain (LBD), and thus are constitutively active. Crucially, they are not responsive to ADT, since these therapies rely on the presence of the LBD. Thus, modulation of AR-V levels using miRs, including variant-specific miRs, may represent a novel therapeutic strategy for mCRPC.
Studies have shown that certain miRs, for example miR-124, are able to target the transcripts of both full-length AR and AR-Vs due to distinct binding sites within the 3’UTRs, and so lead to downregulation of both full length and variant AR.9
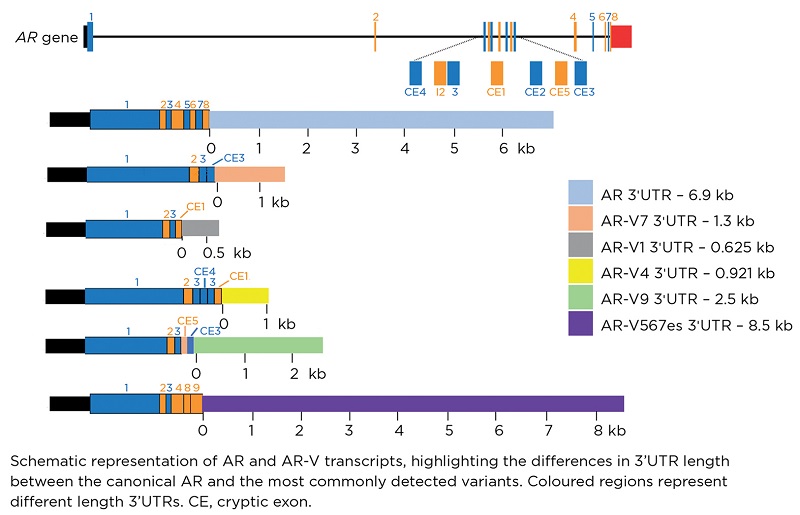
AR-Vs have distinct 3’UTRs when compared with the full length transcript (as shown in the Figure), which is likely to account for the ability of miRs to differently regulate them. The characterisation of these distinct sequences may lead to further identification of additional variant-specific miRs.
THERAPEUTIC POTENTIAL OF MICRORNAs
The challenge for all in the field of cancer research is identifying new therapies that will reduce mortality and morbidity. Excitingly, miRs have several ideal therapeutic properties: they are small, relatively inexpensive to synthesise, highly target-specific, and have the potential to be modified in order to improve in vivo stability and cellular uptake. They also display disease and tissue specificity. Several clinical trials of miR- based therapeutics in cancer have been reported.10,11 The majority utilise synthetic, complementary oligonucleotides which sequester the target miR, inhibiting its function within the cell. Such ‘antagomiRs’ have been reported to be well tolerated, with no long term adverse effects, as seen in phase II trials of anti-miR-122 and anti-miR-155.12
In support of pursuing AR-associated miRs as therapeutic targets in prostate cancer, new evidence is emerging which suggests that aberrant expression of miRs can modulate AR activity, including miRs-221/222. These were identified as upregulated in 90% of mCRPC cases when compared with primary tumours. This altered expression affected how cells are able to respond to androgen treatment, thus suggesting potential involvement of this miR family in the transition to CRPC.13
Dysregulated miRs can also promote cell survival, proliferation and the development of drug resistance. For example, increased miR-21 levels in mCRPC may contribute to docetaxel chemotherapy resistance by downregulating the expression of PDCD4, a known suppressor of tumorigenesis.14
Potentially, the identification of the most critical miR(s) associated with drug resistance will be the cornerstone for the design of therapeutics. To date, antagomiRs and miR mimics are two highly promising miR-based therapeutics. However, it is widely accepted that the effective, targeted delivery of miR therapeutics is still a major challenge that needs to be overcome before these can be used extensively in the clinical setting. With much ongoing research in this area, it can only be a matter of time before miR-based therapies, probably in combination with more traditionally hormone-cased therapies, are used in advanced prostate cancer.
SUE M POWELL, CLAIRE E FLETCHER AND CHARLOTTE L BEVAN
Department of Surgery and Cancer, Imperial College London
ACKNOWLEDGEMENTS
The authors gratefully acknowledge support of their work on miRs and AR by Prostate Cancer UK (RIA17-ST2-017).
REFERENCES
- Hamilton MP et al. 2016 Neoplasia 18 356–370.
- Selth LA et al. 2012 Endocrine-Related Cancer 19 R99–R113.
- Cortez MA et al. 2011 Nature Reviews Clinical Oncology 8 467–477.
- Kumar B et al. 2016 Oncotarget 7 72593–72607.
- Fletcher CE et al. 2019 Oncogene 38 5700–5724.
- Xiao JGA et al. 2012 Prostate 72 1514–1522.
- Nadiminty N et al. 2012 Journal of Biological Chemistry 287 1527–1537.
- Goto Y et al. 2017 British Journal of Cancer 117 409–420.
- Shi XB et al. 2015 Cancer Research 75 5309–5317.
- Reid G et al. 2016 Epigenomics 8 1079–1085.
- van Zandwijk N et al. 2017 Lancet Oncology 18 1386–1396.
- Janssen HL et al. 2013 New England Journal of Medicine 368 1685–1694.
- Sun T et al. 2012 Prostate 72 1093–1103.
- Kopczyńska E 2015 Contemporary Oncology 19 423–427.



{kind=link}